NCS: anemia, carboidratos, catabolismo, anabolismo e glicose
- 10 de jan. de 2021
- 25 min de leitura
1ano 1semestre 3módulo 2SP
06 de Maio de 2021


PROBLEMAS:
Estuda à noite e trabalha intensamente durante o dia ( rotina desgastante)
Sonolento o tempo todo, fraco e desolado
Péssimos hábitos alimentares ( ingere muito carboidrato)
Não ter tempo e vontade para praticar esportes ou fazer exercícios físicos
IMC= 36,8 (obeso II)
Não gosta de hortaliças e frutas
Desequilíbrio entre ingestão e gasto energético
Excesso de calorias
Deposição de gordura ( + abdominal)
Ingestão de alimentos pobres em nutrientes
"Peso aumentado e não praticar exercício é sério condicionante a ter problemas de saúde muito sério"
Obeso II com apenas 16 anos de idade
HIPÓTESES:
Os sintomas de sonolência e fraqueza podem ser decorrentes de uma anemia por déficit de ferro e/ou de vitamina B12
A vitamina C ajuda na absolvição de Fe, protegendo-o da oxidação
Vitamina é uma molécula orgânica que sintetiza proteína no organismo
Carboidratos são cadeias longas de C+H. Ex: glicose, lactase, amido, frutose, etc.
Digestão do carboidrato:
Boca ( amilase salivar) ---> intestino delgado (duodeno) - amilase pancreática (quebra em mono sacarídeo ) ---> absorção no jejuno pelos enterócitos ---> corrente sanguínea --- ação da insulina na célula ---> abertura do canal de glicose ---> mitocôndria ---> respiração celular ---> ATP
6. A respiração celular é composta por 3 etapas: glicólise, C. Krebs e cadeia transportadoras de e-
7. O glicogênio é uma forma de reserva de glicose. Pode ser classificado como hepático ou muscular. O glucagon age no glicogênio hepático
8. O anabolismo se relaciona com a síntese de moléculas estruturais para o corpo e o catabolismo com a quebra de moléculas estruturais. Glucagon - catabolismo Insulina- anabolismo
9. Os neurônios não necessitam de insulina para absorver glicose, diferentemente do tecido esquelético
QUESTÕES:
Conceitue vitamina e como a Vitamina B12 e C se relacionam com anemia
Como a deficiência de ferro pode levar a anemia
Caracterize carboidratos ( o que é e quais são) e explique seu metabolismo desde a ingestão até a formação de ATP
Como o carboidrato pode ser armazenado no corpo humano
Conceitue metabolismo e diferencie anabolismo e catabolismo H -confirmada
Explique de que forma a glicose pode se internalizar nos diferentes tipos celulares I - confirmada
1. Conceitue vitamina e como a Vitamina B12 e C se relacionam com anemia
VITAMINAS
As vitaminas são compostos orgânicos, presentes nos alimentos, essenciais para o funcionamento normal do metabolismo. São essências na transformação de energia, mesmo que não sejam fontes, agem em diferentes sistemas e auxiliam nas respostas imunológicas do organismo, protegendo-o.
As vitaminas são moléculas orgânicas (contendo carbono) que funcionam principalmente como catalisadores para reações dentro do organismo.
As vitaminas são tanto solúveis em gordura como em água.
As solúveis em gordura podem ser lembradas com a sigla mnemônica (que ajuda a memória) ADEK, para as vitaminas A, D, E e K. Essas vitaminas se acumulam dentro da gordura armazenada no organismo e dentro do fígado.
As vitaminas solúveis em água incluem a vitamina C e as vitaminas B. Tanto as vitaminas B como a vitamina C são armazenadas no fígado. As vitaminas são essências na transformação de energia, mesmo que não sejam fontes. Melhoram a pele, a oxigenação das células, auxiliam no funcionamento do metabolismo e ajudam nos processos de cura e rejuvenescimento. A carência de vitaminas no organismo, chamada hipovitaminose ou avitaminose, é responsável pelo surgimento de doenças.
ANEMIA FERROPRIVIA
A anemia é definida como síndrome caracterizada por diminuição de massa eritrocitária total. Laboratorialmente, definimos anemia como hemoglobina menor que 12 g/dl em mulheres ou 13 g/dl em homens. Na gravidez existe anemia relativa, por hemodiluição, além daquela por carência nutricional, principalmente, por deficiência de ferro e ácido fólico. Na gestação os limites considerados normais para o valor da hemoglobina caem para 10g% e os do hematócrito para 30%.
Anemia é definida pela Organização Mundial da Saúde (OMS) como a condição na qual o conteúdo de hemoglobina no sangue está abaixo do normal como resultado da carência de um ou mais nutrientes essenciais
Os principais efeitos adversos e consequências são: fadiga generalizada, anorexia, menor disposição para o trabalho, apatia, retardo no crescimento, perda significativa de habilidade cognitiva, baixo peso ao nascer, mortalidade perinatal e dificuldade de aprendizagem (MINISTÉRIO DA SAÚDE, 2004).

Absorção do ferro
O ferro é absorvido no duodeno e na parte superior do jejuno. A absorção de ferro é determinada pelo tipo de molécula de ferro e de quais outras substâncias são ingeridas. A absorção de ferro é melhor quando o alimento contém ferro-heme (carne). O ferro não heme da dieta deve ser reduzido para o estado ferroso e liberado de aglutinantes alimentares pelas secreções gástricas. A absorção de ferro não heme é reduzida por outros itens alimentares (p. ex., por fitatos de fibras de vegetais e polifenóis; tanatos de chás, incluindo fosfoproteínas; farelo de cereais) e alguns antibióticos (p. ex., tetraciclina). O ácido ascórbico ( vit. C ) é o único elemento alimentício comum que aumenta a absorção de ferro não heme, atuando como componente não oxidante.
ANEMIA MACROCÍTICA
Cobalamina é o termo genérico para componentes com atividade biológica B12. Esses componentes estão envolvidos no metabolismo do ácido nucleico, na transferência de metil e no reparo da síntese de mielina. São necessários para a formação de eritrócitos normais .
Em geral, a anemia se desenvolve de modo insidioso. Ela é mais grave do que os sintomas indicam, pois sua evolução lenta permite adaptação fisiológica. (1)
Anemias megaloblásticas resultam, na maioria das vezes, de deficiências de vitamina B12 e folato. A hematopoese ineficaz afeta todas as linhagens de células, mas, em particular, os eritrócitos. O diagnóstico é normalmente fundamentado em hemograma e esfregaço periférico, que podem mostrar anemia macrocítica com anisocitose e poiquilocitose, grandes eritrócitos ovais (macro-ovalócitos), neutrófilos hipersegmentados e reticulocitopenia. O tratamento é direcionado à causa de base.
Etiologia
A causa mais comum do estado megaloblástico é a deficiência ou utilização defeituosa de vitamina B12 ( Deficiência de vitamina B 12) ou folato ( Deficiência de folato). Outras causas incluem medicamentos (em geral, antineoplásicos ou imunossupressores) que interferem em síntese de DNA e distúrbios metabólicos raros (p. ex., acidúria orótica hereditária); alguns casos têm etiologia desconhecida.
Fisiopatologia
Os estados megaloblásticos resultam da síntese defeituosa de DNA. A síntese de RNA continua, resultando em uma célula maior com núcleo maior. Todas as linhagens celulares têm dispoese, em que a maturidade citoplasmática é maior do que a maturidade nuclear; isso produz megaloblastos na medula antes que apareçam no sangue periférico. A dispoese resulta na morte da célula intramedular, provocando eritropoese ineficaz e causando hiperbilirrubinemia indireta e hiperuricemia. Como a dispoese afeta todas as linhagens celulares, desenvolve-se reticulocitopenia e, durante os estádios tardios, leucopenia e trombocitopenia. Eritrócitos maiores e ovais (macro-ovalócitos) entram na circulação. A hipersegmentação dos neutrófilos polimorfonucleares é comum; o mecanismo de sua produção é desconhecido.
Sinais e sintomas
A anemia se desenvolve de maneira insidiosa e pode não produzir sintomas até se tornar grave. As deficiências de vitamina B12 podem produzir manifestações neurológicas, incluindo neuropatia periférica, demência e degeneração subaguda combinada. A deficiência de folato também pode provocar diarreia e glossite. Muitos pacientes com deficiência de folato parecem fracos, em particular com perda de massa muscular frontal.
(2)
NOVA SÍNTESE:
REFERÊNCIAS:
(1) Vitamina B 12 (Cobalaminas)
Por Larry E. Johnson, MD, PhD, Associate Professor of Geriatrics and Family and Preventive Medicine, University of Arkansas for Medical Sciences; Medical Director, Central Arkansas Veterans Healthcare System
(2) Anemias macrocíticas megaloblásticas
Por Alan E. Lichtin, MD, Associate Professor, Cleveland Clinic Lerner College of Medicine; Staff Hematologist-Oncologist, Cleveland Clinic
2. Como a deficiência de ferro pode levar a anemia A,B
---ANEMIA MICROCÍTICA/FERROPRIVA
O ferro desempenha importantes funções no metabolismo humano, tais como transporte e armazenamento de oxigênio, reações de liberação de energia na cadeia de transporte de elétrons, conversão de ribose a desoxirribose, co-fator de algumas reações enzimáticas e inúmeras outras reações metabólicas essenciais. Normalmente, cerca de 70 a 90% do ferro é captado pela medula óssea para ser utilizado na produção da hemoglobina. O ferro é estocado no fígado, sob as formas de ferritina e hemossiderina.
A anemia é o último estágio da deficiência de ferro. O primeiro estágio, depleção de ferro, afeta os depósitos e representa um período de maior vulnerabilidade em relação ao balanço marginal de ferro, podendo progredir até uma deficiência mais grave, com consequências funcionais. Neste estágio há diminuição da ferritina sérica.
O segundo estágio, deficiência de ferro, é referido como uma eritropoiese ferro-deficiente e caracteriza-se por alterações bioquímicas que refletem a insuficiência de ferro para produção normal de hemoglobina e outros compostos férricos, ainda que a concentração de hemoglobina não esteja reduzida. Neste estágio há declínio da concentração de ferro sérico e aumento na capacidade de ligação de ferro.
O terceiro e último estágio, anemia ferropriva, caracteriza-se pela diminuição dos níveis de hemoglobina, com prejuízos funcionais ao organismo, tanto mais graves quanto maior for essa redução. A redução na concentração de hemoglobina presente neste estágio é o parâmetro universal para definir anemia.
Absorção do ferro
O ferro é absorvido no duodeno e na parte superior do jejuno. A absorção de ferro é determinada pelo tipo de molécula de ferro e de quais outras substâncias são ingeridas. A absorção de ferro é melhor quando o alimento contém ferro-heme (carne). O ferro não heme da dieta deve ser reduzido para o estado ferroso e liberado de aglutinantes alimentares pelas secreções gástricas. A absorção de ferro não heme é reduzida por outros itens alimentares (p. ex., por fitatos de fibras de vegetais e polifenóis; tanatos de chás, incluindo fosfoproteínas; farelo de cereais) e alguns antibióticos (p. ex., tetraciclina). O ácido ascórbico ( vit. C ) é o único elemento alimentício comum que aumenta a absorção de ferro não heme.
Transporte e uso do ferro
O ferro das células da mucosa intestinal é transferido para a transferrina, uma proteína transportadora de ferro sintetizada no fígado; a transferrina pode transportar o ferro das células (intestinal, macrófagos) aos receptores específicos em eritroblastos, células da placenta e células do fígado. Para a síntese de heme, a transferrina transporta ferro para as mitocôndrias do eritroblasto, que inserem o ferro na protoporfirina, para que esta se transforme em heme. A transferrina (meia-vida plasmática de oito dias) é expelida para reutilização. A síntese de transferrina aumenta com a deficiência de ferro, mas diminui com qualquer tipo de doença crônica.
Armazenamento e reciclagem de ferro
O ferro não utilizado na eritropoese é transferido pela transferrina, uma proteína de transporte de ferro, para o pool de armazenamento; o ferro é armazenado de duas formas, ferritina e hemossiderina. A mais importante é a ferritina (um grupo heterogêneo de proteínas ao redor de um núcleo de ferro), que é a fração solúvel e ativa de depósito localizado no fígado (em hepatócitos), na medula óssea e no baço (em macrófagos); nos eritrócitos; e no soro. O ferro armazenado na ferritina está prontamente disponível para qualquer necessidade do corpo. Os níveis séricos de ferritina traçam um paralelo com o tamanho dos depósitos corporais (1 ng/mL = 8 mg de ferro no pool de armazenamento). O segundo pool de armazenamento do ferro é a hemossiderina, que é relativamente insolúvel e armazenada primariamente no fígado (nas células de Kupffer) e na medula óssea (nos macrófagos).
Como a absorção de ferro é muito limitada, o corpo recicla e conserva o ferro. A transferrina segura e recicla o ferro disponível dos eritrócitos senescentes, sujeitando-os à fagocitose pelos fagócitos mononucleares. Esse mecanismo fornece cerca de 97% do ferro diário necessário (em torno de 25 mg de ferro). Com o envelhecimento, os depósitos de ferro tendem a aumentar porque sua eliminação é lenta.
Deficiência de ferro
A deficiência se desenvolve em estágios.
No primeiro estágio, a necessidade de ferro excede sua ingestão, causando depleção progressiva dos depósitos de ferro da medula óssea. À medida que os estoques diminuem, há elevação da absorção dietética de ferro. Durante os estágios tardios, a deficiência é grave o suficiente para ocasionar insuficiência na síntese de eritrócitos, gerando anemia.
A deficiência de ferro, se for grave e prolongada, também pode provocar disfunção no conteúdo de ferro nas enzimas celulares.
Etiologia
Como o ferro é mal absorvido, a maioria das pessoas mal alcança sua necessidade diária. Mesmo assim, as pessoas que têm dieta tipicamente ocidental têm menos probabilidade de ter deficiência de ferro unicamente como resultado de deficiência dietética. Assim, mesmo com perdas modestas, o aumento das necessidades ou a diminuição de sua ingestão rapidamente produz a deficiência de ferro.
Perda de sangue é quase sempre a causa. Em homens, a origem mais frequente é o sangramento crônico oculto, muitas vezes do trato gastrintestinal (GI). Em mulheres em pré-menopausa, a perda menstrual cumulativa de sangue (o que significa 0,5 mg ferro/dia) é a causa comum. Outra possível causa da perda de sangue em homens e mulheres é hemólise intravascular crônica ( Visão geral da anemia hemolítica) quando a quantidade de ferro liberada durante a hemólise excede a capacidade de ligação à haptoglobina. A deficiência de vitamina C pode contribuir para a anemia ferropriva causando fragilidade capilar, hemólise e sangramento.
Aumento da necessidade de ferro pode contribuir para a deficiência de ferro. Do nascimento até dois anos de idade e durante a adolescência, quando o rápido crescimento requer grande ingestão de ferro, a dieta de ferro, muitas vezes, não é adequada. Durante a gestação, a necessidade de ferro fetal eleva a necessidade de ferro materno (em média 0,5 a 0,8 mg/dia — Anemia na gestação), mesmo com a ausência das menstruações. A lactação também aumenta a necessidade de ferro (em média 0,4 mg/dia).
Diminuição da absorção de ferro pode resultar de gastrectomia e síndromes de má absorção do intestino delgado superior. Raramente, a absorção diminui pela privação da dieta advinda de má nutrição.
Estágios da deficiência de ferro
Resultados de testes laboratoriais ajudam a determinar o estágio da anemia por deficiência de ferro.
O estádio 1 caracteriza-se pela diminuição dos depósitos de ferro na medula óssea; Hb e ferro sérico permanecem normais, mas a concentração de ferritina sérica cai para < 20 ng/mL. O aumento compensatório da absorção do ferro causa elevação da capacidade de ligação do ferro (nível de transferrina).
Durante o estádio 2, a eritropoese é insuficiente. Embora o nível de transferrina seja aumentado, a concentração do ferro sérico diminui; a saturação da transferrina reduz. A eritropoese é insuficiente quando o ferro sérico cai para abaixo de 50 μg/dL (< 9 μmol/L) e a saturação de transferrina para < 16%. A concentração do receptor de ferritina sérica aumenta (> 8,5 mg/dL).
Durante o estádio 3, a anemia com aparecimento de eritrócitos normais e índices normais se desenvolvem.
Ao longo do estádio 4, desenvolve-se microcitose e, em seguida, hipocromia.
Durante o estádio 5, a deficiência de ferro afeta os tecidos, resultando em sinais e sintomas.
O diagnóstico da anemia por deficiência de ferro tem de levar em consideração a causa, que normalmente consiste no sangramento. Os pacientes com perdas de sangue óbvias (p. ex., mulheres com menorragia) podem não precisar de exames posteriores. Homens e mulheres em pós-menopausa sem perda de sangue óbvia devem se submeter à avaliação do trato GI, porque a anemia pode ser apenas uma indicação de câncer GI oculto. Raramente, a epistaxe crônica ou o sangramento geniturinário (GU) é subestimado pelo paciente e requer avaliação em pacientes com resultados normais dos exames GI.
NOVA SINTESE:
REFERÊNCIAS:
Anemia por deficiência de ferro (Anemia por perda crônica de sangue; clorose)
Por Alan E. Lichtin, MD, Associate Professor, Cleveland Clinic Lerner College of Medicine; Staff Hematologist-Oncologist, Cleveland Clinic
3. Caracterize carboidratos ( o que é e quais são) e explique seu metabolismo desde a ingestão até a formação de ATP D - confirmada parcialmente, E - confirmada parcialmente, F - confirmada parcialmente
Carboidratos são poliidroxialdeídos ou poliidroxicetonas ou substâncias que liberam tais compostos por hidrólise.
O termo carboidratos denota hidratos de carbono, designação oriunda da fórmula geral (CH2 O)n apresentada pela maioria dessas moléculas. Podem ser divididos em três classes principais de acordo com o número de ligações glicosídicas: monossacarídeos, oligossacarídeos e polissacarídeos (1)
Eles possuem grande variedade de funções, que incluem o fornecimento de fração significativa da energia na dieta da maioria dos organismos e a atuação como forma de armazenamento de energia no corpo e como componentes da membrana celular, mediando algumas formas de comunicação intercelular. (2)
Os monossacarídeos consistem somente de uma unidade de poliidroxialdeídos ou cetonas, as quais podem ter de três a sete átomos de carbono. Devido à alta polaridade, são sólidos cristalinos em temperatura ambiente, solúveis em água e insolúveis em solventes não polares. Suas estruturas são configuradas por uma cadeia carbônica não ramificada, na qual um dos átomos de carbono é unido por meio de uma dupla ligação a um átomo de oxigênio, constituindo assim um grupo carbonila. O restante dos átomos de carbono possui um grupo hidroxila (daí a denominação de poliidroxi). Quando o grupo carbonila está na extremidade da cadeia, o monossacarídeo é uma aldose. Caso o grupo carbonila esteja em outra posição, o monossacarídeo é uma cetose.
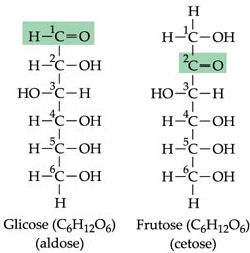
Os oligossacarídeos são formados por cadeias curtas de monossacarídeos geralmente são formados por 3 até 10 unidades de monossacarídeo.. Os mais comuns são os dissacarídeos, dos quais se destacam a sacarose (açúcar da cana) e a lactose (açúcar do leite)

Polissacarídeos Açúcares contendo mais de 20 unidades são denominados polissacarídeos, os quais podem possuir milhares de monossacarídeos e são a forma predominante dos carboidratos na natureza. A diferenciação é dada pela unidade monomérica, comprimento e ramificação das cadeias. Quando os polissacarídeos contêm apenas um tipo de monossacarídeo, ele é denominado de homopolissacarídeo. Se estiverem presentes dois ou mais tipos de monossacarídeos, o resultado é um heteropolissacarídeo. Ex: celulose, glicogênio não ramificado e amido.
A digestão humana é extracelular, pois ocorre no interior do tubo digestivo. Compreende processos físicos (mecânicos) como a mastigação, a deglutição e os movimentos peristálticos. É também um processo químico, graças à ação das enzimas secretadas por glândulas anexas. O processo digestivo inicia-se na boca pela ação trituradora dos dentes.
A mastigação é a primeira etapa do processo digestivo nos animais que possuem dentes, uma etapa mecânica e o ato de engolir (deglutição), também mecânico, ocorre graças ao músculo, revestido de tecido conjuntivo conhecido como língua. A língua tem sua extremidade posterior presa ao osso hióide. Desempenha importante papel na percepção do gosto, pois nela estão localizadas as papilas gustativas. Tem papel importantíssimo também na fonação. É inervada por dois pares de nervos cranianos: glossofaríngeo e o hipoglosso. Mantém-se constantemente umedecida pela secreção das glândulas salivares. Da língua o bolo alimentar é deglutido para a faringe que através de movimentos voluntários levam o bolo alimentar para o esôfago. Do esôfago, através de contrações involuntárias o alimento chega ao estômago.
A faringe e a parte anterior do esôfago apresentam músculos estriados (voluntários). A parte posterior do esôfago, o estômago e o intestino, possuem musculatura lisa (involuntária). O alimento transita ao longo do tubo digestivo, graças aos movimentos peristálticos. A musculatura lisa do tubo digestivo é inervada pelo sistema nervoso autônomo (simpático e parassimpático). A estimulação do parassimpático aumenta o peristaltismo da musculatura lisa gastrointestinal, enquanto que a estimulação do simpático a modera ou inibe completamente. Concomitantemente ao "trânsito" do bolo alimentar através do tubo digestivo, ocorre a digestão química dos alimentos com a subseqüente absorção dos componentes digeridos. No final deste processo os "restos" dos alimentos ingeridos que não foram degradados, que conhecemos como fezes, são armazenados no ceco, para posteriormente serem eliminados pelo ato involuntário da defecação.
ETAPAS DA DIGESTÃO QUÍMICA
Os processos químicos constituem a transformação das grandes moléculas de proteínas, lipídios, glicídios e ácidos nucléicos em pequenas moléculas que serão absorvidas para corrente sanguínea através da mucosa intestinal. Neste processo intervêm as enzimas que são secretadas pelas glândulas anexas ao tubo digestivo.
NA BOCA:
deve-se à ação de enzimas da saliva que é secretada pelas glândulas salivares parótidas, submaxilares, sublinguais e em outras glândulas salivares menores. A principal enzima da saliva é a amilase salivar (ptialina). Outras enzimas presente na saliva como a maltase e catalase são de menor importância porque são produzidas em quantidades menores. A saliva tem pH entre 6,4 - 7,5, que favorece a ação da amilase salivar. Esta catalisa a hidrólise de polissacarídeos (amido, glicogênio e seus derivados). A digestão do amido (polissacarídeo) pela saliva produz oligossacarídeos e maltose. Quando o alimento é colocado na boca, reflexos nervosos estimulam a secreção da saliva, especialmente se o alimento é saboroso ou apetitoso. Tal controle é realizado pelo sistema nervoso autônomo. O SNP estimula secreção e o SNS inibe a secreção.

DIGESTÃO NO ESTÔMAGO:
no estômago o alimento sofre a ação do suco gástrico que é secretado pelas glândulas localizadas na parede estomacal. O muco é produzido pelas glândulas pilóricas e cárdicas do estômago e lubrifica o bolo alimentar, além de proteger a parede do estômago contra a ação das enzimas gástricas e do HCl.
O HCl apresenta as seguintes funções: facilita a absorção de ferro; proporciona um pH ótimo para a digestão protéica; ativa o Pepsinogênio à Pepsina; age contra os germes restringindo a fermentação microbiana (ação germicida).
As enzimas do suco gástrico são: pepsina, lípase gástrica, amilase gástrica.
A pepsina é uma enzima proteolítica (digere proteínas em peptídeos), que atua num meio altamente ácido (pH = 2,0) e acima de pH = 5,0 apresenta pouca atividade proteolítica, tornando-se inativa.
A lípase gástrica (tributirase) age sobre a tributirina (um tipo de gordura encontrado no leite e seus derivados), quase não tem atividade lipolítica sobre as gorduras comuns.
A amilase gástrica não desempenha papel importante na digestão do amido. A secreção gástrica é regulada por mecanismos nervosos e hormonais. A regulação hormonal é realizada por meio de dois hormônios: gastrina e enterogastrona. A gastrina é produzida pela mucosa da região pilórica do próprio estômago e tem ação estimulante sobre a secreção gástrica (fig.7.3). A enterogastrona é produzida no intestino delgado (duodeno) em presença de gordura e inibe a secreção gástrica.
DIGESTÃO NO INTESTINO:
As enzimas encontradas no intestino delgado decorrem do suco pancreático, secretado por um órgão anexo ao aparelho digestivo, o pâncreas.
Suco pancreático: é secretado pelo pâncreas (parte exócrina), seu pH é de 7,8 - 8,2 devido ao alto teor em bicarbonato. As enzimas desse suco são: Tripsina, quimotripsina, carboxi e amino-peptidase, amilase pancreática, lípase pancreática, ribonuclease e desoxirribonuclease.
TRIPSINA: é sintetizada nas células pancreáticas na forma do precursor inativo (tripsinogênio). A ativação do tripsinogênio é, realizada pela enzima enteroquinase (produzida pelo intestino delgado). O tripsinogênio também pode ser ativado pela própria tripsina (autocatálise). Esta enzima atua sobre proteínas inteiras ou parcialmente digeridas produzindo frações menores (peptídeos).
QUIMOTRIPSINA: é produzida pelo pâncreas na forma de quimotripsinogênio que é ativado pela tripsina, passando, então a quimotripsina. Esta enzima age sobre proteínas inteiras ou parcialmete digeridas produzindo frações menores (peptídeos).
CARBOXI e AMINO PEPTIDASE: digerem peptídeos a aminoácidos pela região carboxi e amino terminal, respectivamente.
AMILASE PANCREÁTICA: hidrolisa os polissacarídeos a dissacarídeos. OBS: Alguns polissacarídeos, como a celulose e a quitina, não são hidrolisados pelas amilases humanas. LIPASE PANCREÁTICA: hidrolisa as gorduras neutras, ácidos graxos e glicerol.
NUCLEASES: (ribonuclease e desoxirribonuclease) hidrolisam, respectivamente, o ácido ribonucléico e o desoxirribonucléico a frações menores (nucleotídeos).
A secreção pancreática é regulada por mecanismo nervoso e também hormonal.
A visão, o cheiro, o paladar e também a chegada do bolo alimentar ao estômago desencadeiam impulsos parassimpáticos através do nervo vago até o pâncreas, determinando uma secreção moderada do suco pancreático.
A chegada do alimento ao intestino delgado estimula a mucosa duodenal a produzir os hormônios secretina e pancreosina, que, por sua vez, estimulam o pâncreas a secretar o suco pancreático.
A secretina é produzida em resposta à estimulação da acidez do bolo alimentar que chega ao intestino delgado. O suco pancreático, que chega no duodeno, é altamente rico em bicarbonato que tem por finalidade neutralizar a acidez do bolo alimentar e, assim, garantir a ação das enzimas pancreáticas que funcionam em pH ligeiramente alcalino e neutro.
Outro anexo do aparelho digestivo é a vesícula biliar que armazena um líquido denominado Bile.
A BILE emulsifica as gorduras, é produzida pelo fígado a partir de hemácias velhas e é armazenada na vesícula biliar. Não apresenta enzimas digestivas. Possui sais biliares (glicolato e taurocolato de sódio) que emulsionam as gorduras, facilitando a ação das lípases (aumentam a superfície de ação). Outra função dos sais biliares é solubilizar os produtos finais da digestão lipídica, facilitando assim a sua absorção através da mucosa intestinal. A presença de gordura no intestino delgado estimula a mucosa duodenal a produzir o hormônio colecistoquinina, o qual age determinando a contração da parede da vesícula que, então, elimina a bile para o intestino.
Em sua maior parte os sais biliares são reabsorvidos pelo intestino e a seguir reutilizados pelo fígado várias vezes, antes de serem transformados em biliverdina (pigmento que da a cor às fezes).
SUCO ENTÉRICO: é produzido pelo epitélio glandular das criptas de Lieberkuhen, localizadas no intestino delgado.
O suco entérico (intestinal) contém muco, cuja função é proteger a parede intestinal contra uma autodigestão, e as enzimas: enteroquinase, erepsina e as enzimas produzidas pelo pâncreas: lípase, amilase, maltase, lactase e sucrase. Seu pH está na faixa de 6,5 a 7,5
A enteroquinase, além do papel de ativadora do tripsinogênio, digere peptídeos a aminoácidos.
Importantes estímulos diretos ou reflexos regulam a secreção do intestino delgado. A distensão do intestino e estímulos táteis ou irritantes resultam em intensa secreção do suco intestinal. A secretina um dos principais hormônios produzidos pelo intestino delgado, tem ação sobre as células do ducto pancreático e do trato biliar, aumentando a secreção de bicarbonato, o que produz um suco pancreático aquoso alcalino.


ABSORÇÃO DOS ALIMENTOS:
A absorção dos alimentos ocorre principalmente no intestino delgado, que possui microvilosidades, estruturas responsáveis pelo aumento da superfície de absorção. Ao nível do jejuno-íleo há uma grande absorção de glicose, aminoácidos, etc. O estômago e o intestino grosso também participam da absorção, principalmente de água. Algumas substâncias são absorvidas por pinocitose, porém a maior parte da absorção ocorre por difusão e transporte ativo. Uma população bacteriana está presente no intestino grosso, sendo responsável pela produção de vitaminas: k, B12, tianina, riboflavina e vários gases.
O catabolismo inicia com a digestão, onde moléculas de polissacarídeos são quebradas em monosacarídeos, lipídios são quebrados em ácidos graxos e glicerol e proteínas são quebradas em seus aminoácidos. Após a digestão temos os catabolismos de carboidratos, de lipídios e de aminoácidos, que iremos estudar nesta e nas duas próximas aulas. Todos esses três metabolismos convergem para o Ciclo do Ácido Cítrico, também denominado de Ciclo de Krebs, onde a acetil-CoA é convertida em CO2 e H2 O, e a energia é conservada na forma de ATP, GTP, NADH e FADH2 . Por fim temos a etapa da fosforilação oxidativa onde todos são convertidos em ATP, armazenando assim a energia.
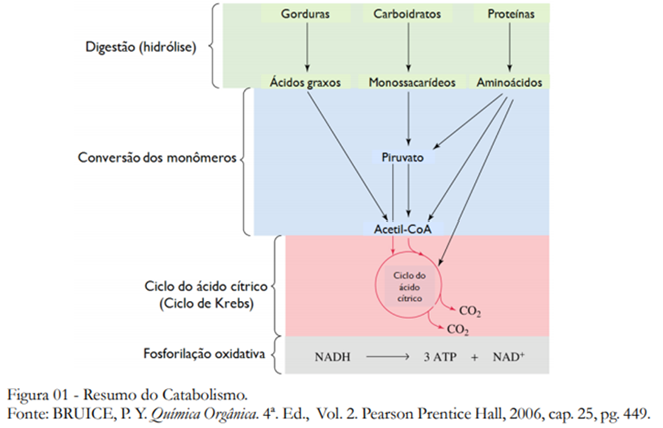
CATABOLISMO DE CARBOIDRATOS
Os carboidratos possuem um monômero centralisador que é a glicose. Da glicose os carboidratos podem ser estocados na forma de glicogênio nos animais, ou na forma de amido ou sacarose nas plantas. Quando o corpo necessita de energia é acionada a via glicolítica, onde a glicose é convertida a piruvato. Por fim, partindo da glicose sintetizamos as pentoses necessárias na síntese de ácidos nucleicos, pela via das pentoses.
GLICÓLISE
A glicólise é o catabolismo da glicose. Nela a molécula de 6 carbonos da glicose é convertida em duas moléculas de 3 carbonos de piruvato, gerando energia na forma de ATP e NADH durante o processo. A glicólise pode ser dividida em duas etapas de 5 reações cada.
A primeira etapa é denominada de fase preparatória, e nesta etapa ATP é consumido para ativar a molécula de glicose e convertê-la em gliceraldeído-3-fosfato (Figura 3).

Na etapa seguinte, denominada de fase de compensação, o gliceraldeído3-fosfato é convertido em piruvato, e energia é armazenada na forma de ATP e NADH (Figura 4).

A glicólise inicia com a fosforilação da glicose. Nesta etapa a enzima hexoquinase utiliza uma molécula de ATP para converter a glicose em glicose-6-fosfato (Figura 5). O mecanismo envolve um ataque nucleofílico da hidroxila do álcool primário na posição C-6 ao fósforo-γ do ATP. O ADP é o grupo abandonador. Embora essa reação seja energeticamente favorável, com ΔGº = - 16,7 kJ/mol, ela não ocorreria sem o auxílio da enzima e do Mg2+, pois as cargas negativas dos grupos fosfato impediriam a aproximação do nucleófilo na substituição nucleofílica (Figura 6). Nota que todos os mecanismos envolvendo o ATO serão similares, não havendo a necessidade deles serem mostrados novamente.


Na segunda reação da glicólise, a enzima fosfo-hexose-isomerase, através de quatro reações sucessivas de catálise enzimática básica geral e ácida geral, promove a conversão de glicose-6-fosfato em frutose-6-fosfato (Figura 7). A primeira etapa do mecanismo envolve o ataque de um resíduo básico de Glu à hidroxila do carbono anomérico, abrindo a ligação hemiacetal da glicose-6-fosfato. Em seguida outro resíduo básico de Glu remove o Hα carbonílico que possui caráter ácido, formando assim o ¬cisenediol. Um resíduo ácido de Glu doa o H ácido ao enediol, isomerizando a glicose-6-fosfato para frutose-6-fosfato, e por fim ocorre o fechamento do anel numa ligação hemicetal.

Na terceira reação da glicólise, a enzima fosfofrutoquinase catalisa a transferência de um fosfato do ATP para a hidroxila do C-1 da frutose6-fosfato, formando a frutose-1,6-bifosfato (Figura 9). O mecanismo é análogo ao da Figura 6.


Na quarta reação da glicólise, a enzima aldolase catalise a reversão de uma condensação aldólica, convertendo a frutose-1,6-bifosfato em gliceraldeído-3-fosfato e diidroxiacetona-fosfato (Figura 10). O mecanismo envolve o ataque nucleofílico de um resíduo de Lys à carbonila cetônica da frutose-1,6-difosfato, formando uma base de Schiff protonada na enzima. O deslocamento eletrônico facilita a etapa seguinte que envolve a captura do hidrogênio da hidroxila do C-4, levando à clivagem da ligação C3-C4 revertendo a condensação aldólica e liberando o primeiro produto, o gliceraldeído-3-fosfato. O enol da imina então tautomeriza formando nova base de Schiff protonada, que hidrolisa restaurando a enzima e liberando a diidroxiacetona-fosfato (Figura 11).
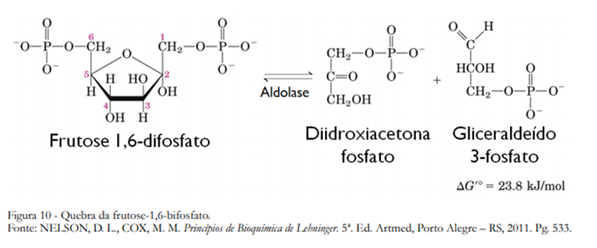

Na quinta reação da glicólise, e última da fase preparatória, a diidroxiacetona-fosfato é isomerizada à gliceraldeído-3-fosfato (Figura 12), num mecanismo análogo ao da isomerização da glicose-6-fosfato à frutose-6-fosfato (Figura 8). A diferença é o número menor de etapas pois a estrutura já se encontra aberta, não havendo a necessidade de quebra da ligação hemiacetal e nem da formação da ligação hemicetal.

Na sexta reação da glicólise entramos na fase compensação. Até o momento gastamos duas moléculas de ATP para converter a glicose em duas moléculas de gliceraldeído-3-fosfato. Agora chegou a hora de vermos nosso investimento dar retorno. A enzima gliceraldeído-3-fosfato-desidrogenase utiliza o NAD+ para oxidar o gliceraldeído-3-fosfato e incorporar em sua molécula um fosfato inorgânico, gerando o 1,3-bifosfoglicerato (Figura 13). Inicialmente o grupo aldeído sofre um ataque nucleofílico do grupo tiol (-SH) de um resíduo de Cys, formando um tio-hemiacetal e se ligando a enzima. Em seguida o NAD+ captura o hidrogênio do C-1, enquanto um resíduo básico captura o hidrogênio da hidroxila, convertendo o tiohemiacetal em tio-éster e liberando uma molécula de NADH. A carbonila então sofre ataque nucleofílico do fosfato inorgânico, numa substituição acílica nucleofílica, gerando o 1,3-bifosfoglicerato e regenerando a enzima (Figura 14).

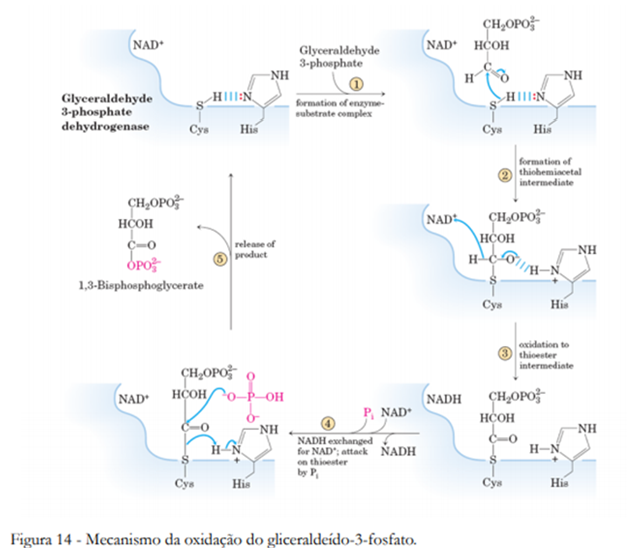
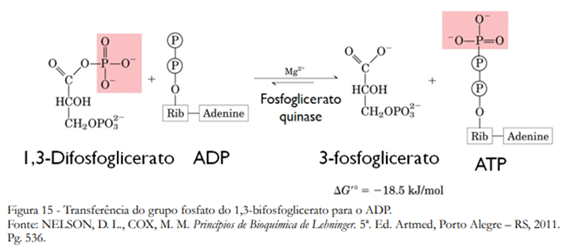
Na sétima etapa da glicólise, o grupo fosforil do 1,3-bifosfoglicerato é transferido para uma molécula de ADP pela enzima fosfoglicerato-quinase, formando ATP e 3-fosfoglicerato (Figura 15). O mecanismo é análogo em todas as fosforilações, mas dessa vez o sentido energeticamente favorável é a transferência do fosfato para o ADP.
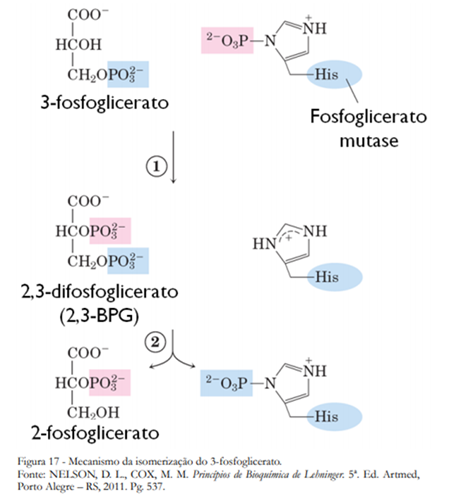
Na oitava etapa da glicólise a enzima fosfoglicerato-mutase catalisa a isomerização do 3-fosfoglicerato em 2-fosfoglicerato (Figura 16). Inicialmente o grupo fosforil é transferido da His para a hidroxila em C-2 do 3-fosfoglicerato, formando assim o 2,3-bifosfoglicerato. O grupo fosforil é então transferido do C-3 para o mesmo resíduo de His produzindo o 2-fosfoglicerato e regenerando a enzima. O Mg2+ exerce papel fundamental nesta reação, neutralizando as cargas negativas dos fosfatos (Figura 17).

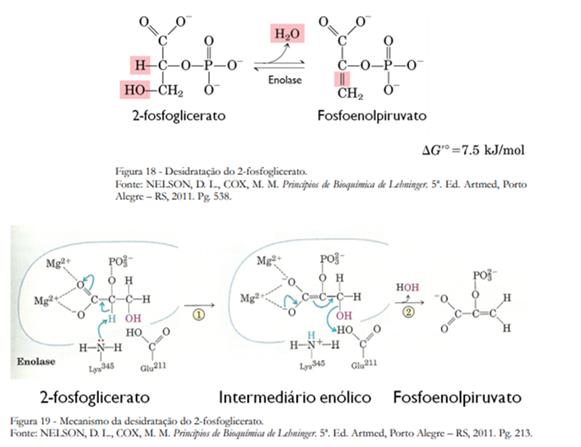
Na nona etapa da glicólise a enzima enolase desidrata o 2-fosfoglicerato, formando o fosfoenolpiruvato (Figura 18). Um resíduo de Lys captura o H ligado ao C-2, sendo que o enol é estabilizado por dois íons Mg2+. Em seguida o Glu facilita a saída da água por uma catálise ácida, formando o fosfoenolpiruvato (Figura 19).
Na décima e última etapa da glicólise o fosfoenolpiruvato transfere seu fosfato para o ADP, gerando o enol do piruvato que tautomeriza para piruvato, e uma molécula de ATP (Figura 20).


Como podemos notar, há um ganho energético na glicólise de 2ATP. Além disso, os elétrons conservados na forma de NADH serão utilizados na fosforilação oxidativa para a conversão de ADP em ATP.
http://www.cesadufs.com.br/ORBI/public/uploadCatalago/12285710072012Quimica_Biomoleculas_aula_11.pdf
REFERÊNCIAS:
(2) Livro: Bio química ilustrada -
(1) Conceitos Científicos em Destaque, Carboidratos: Estrutura, Propriedades e Funções, 28º Encontro de Debates dobre o Ensino de Química , 2008 - http://qnesc.sbq.org.br/online/qnesc29/03-CCD-2907.pdf
4. Como o carboidrato pode ser armazenado no corpo humano G - confirmada
Os alimentos energéticos como carboidratos, gorduras e proteínas podem ser oxidados nas células e neste processo uma grande quantidade de energia será liberada. Essa energia derivada da oxidação é usada para converter o difosfato de adenosina (ADP) em trifosfato de adenosina (ATP), que é largamente utilizada por nosso organismo: no transporte ativo através das membranas biológicas, nas diversas reações de síntese de moléculas essenciais para nosso sistema biológico, na contração muscular, no impulso nervoso e em muitas outras funções primordiais para a manutenção da homeostase.
A regulação do metabolismo da glicose (carboidrato) é feita por dois hormônios peptídicos secretados pelo pâncreas, a insulina e o glucagon. As células endócrinas do pâncreas estão em aglomerados, chamados ilhotas de Langerhans. Existem, aproximadamente, um milhão dessas ilhotas neste órgão, cada uma contendo perto de 2.500 células.
A insulina, liberada sempre em estado alimentado, é chamada de hormônio hipoglicemiante, pois quando a glicose é absorvida pelo intestino, a insulina garante que esse nutriente seja armazenado como glicogênio no figado e no músculo esquelético e como lipídio (quando em excesso) no tecido adiposo. Estas formas de armazenamento podem se tornar disponíveis durante os períodos de jejum. A insulina provoca diminuição da concentração sanguínea de glicose, limitando o aumento sanguíneo desta após a ingestão de carboidratos.
O resultado da ação hipoglicemiante da insulina são respostas que estimulam a oxidação da glicose e inibem a gliconeogênese. Essas ações são coordenadas e simultâneas. Nelas, a insulina promove a glicogênese a partir da glicose (no figado e no músculo esquelético) e inibe a glicogenólise, além de inibir a gliconeogênese e também aumentar o transporte de glicose para as células (músculo esquelético e tecido adiposo) com auxílio de transportadores de glicose (GLUT) nas membranas celulares.
O outro hormônio sintetizado e secretado pelas células das ilhotas de Langerhans é o glucagon. Ao contrário da insulina, ele é liberado sempre nos períodos de jejum, e é considerado o hormônio hiperglicemiante, pois promove a utilização das reservas e não o armazenamento, como a insulina. As ações do glucagon são para aumentar e manter a concentração sanguínea de glicose. O principal sinal regulatório da secreção do glucagon e da insulina é o nível de glicemia, sendo que predomina a liberação de glucagon em momentos de diminuição de glicose sanguínea, enquanto há uma maior liberação de insulina quando a glicemia aumenta.
Referência:
5. Conceitue metabolismo e diferencie anabolismo e catabolismo H -confirmada
Metabolismo é o conjunto de reações químicas que ocorrem nas células e que lhe permitem manter-se viva, crescer e dividir-se. O metabolismo divide-se classicamente em:
• Catabolismo - obtenção de energia e poder redutor a partir de macromoléculas como proteínas, triacilgliceróis.
• Anabolismo - produção de novos componentes celulares, em processos que geralmente utilizam a energia e o poder redutor a partir de moléculas menores como aminoácidos.
O metabolismo celular é o conjunto de reações que ocorrem no ambiente celular com o objetivo de sintetizar as biomoléculas ou degradá-las para produzir energia. O metabolismo de síntese das biomoléculas é conhecido como anabólico (anabolismo) e o de degradação catabólico (catabolismo). O anabolismo ocorre quando a célula dispõe de energia ou substrato suficiente. O catabolismo, por sua vez, ocorre em situações em que o organismo necessita de energia como, por exemplo, entre as refeições e no jejum. O catabolismo produzirá energia na forma de ATP quando as biomoléculas forem degradadas. Como visto na aula de ácidos nucléicos o ATP é um nucleotídeo cuja função é participar das reações de transferência de energia da célula, daí o porquê ser conhecido como a moeda energética da célula. As reações do anabolismo e do catabolismo são opostas, mas que ocorrem de maneira articulada, permitindo a maximização da energia disponível. Assim, enquanto o catabolismo ocorre de maneira espontânea, reação exergônica, com produção de ATP o anabolismo é não espontâneo, ou endergônico, necessitando energia para ocorrer. As biomoléculas energéticas são os carboidratos, lipídios e proteínas que são obtidas em grandes quantidades durante a alimentação ou são mobilizadas das reservas orgânicas quando são ingeridas em quantidade insuficiente na alimentação ou quando o consumo energético aumenta grandemente (p.ex.: durante a realização de exercícios físicos). A forma final de absorção da energia contida nessas moléculas se dá na forma de ligações de alta energia do ATP o qual é sintetizado nas mitocôndrias por processos oxidativos que utilizam diretamente o O2 . Desta forma, é essencial a presença de mitocôndrias e de oxigênio celular para o aproveitamento energético completo das biomoléculas.
Referência:
6. Explique de que forma a glicose pode se internalizar nos diferentes tipos celulares I - confirmada
A glicose, principal fonte de energia celular, é transportada na maioria das células por difusão facilitada, através de proteínas transportadoras presentes na membrana plasmática. Está caracterizada a existencia de urna familia de transportadores (GLUT1-GLUT7), com características funcionáis e distribuição tecidual distintas. Por outro lado, em epitelios intestinal e tubular renal, o transporte é contra gradiente e acoplado ao Na+ na membrana apical das células através de cotransportadores (SGLT1-SGLT2), com posterior difusão para o intersticio através de GLUTs presentes na membrana basolateral.
A glicose é uma molécula polar, insolúvel na membrana plasmática, e o seu transporte é realizado através de difusão facilitada, portanto a favor de seu gradiente de concentração, e dependente da presença de proteínas transportadoras (GLUTs) na superfície de todas as células. Além disso, em células epiteliais como as do intestino delgado e do túbulo renal, os processos de absorção e reabsorção respectivamente, ocorrem através de um processo de transporte acoplado ao íon sódio, o qual promove um transporte contra gradiente de concentração de glicose e a favor do gradiente de concentração de Na+, através de proteínas transportadoras (SGLTs) presentes no bordo em escova da célula epitelial. Nestas células, a glicose concentrada no intracelular difunde-se para o extracelular por difusão facilitada através de GLUTs presentes na membrana basolateral.
Os GLUTs têm capacidade de realizar fluxo bidirecional de glicose e, de fato, é o gradiente do substrato que determinará a direção intra ou extracelular da glicose (1). Considerando-se que a glicose, como substrato energético, está constantemente sendo consumida nas células, as forças de gradiente garantem um influxo do substrato na maioria dos tipos celulares, através das diferentes isoformas de transportadores. Entretanto, basta a concentração intracelular de glicose ser maior que a extracelular para que as forças de gradiente promovam um efluxo do substrato através da isoforma presente. Isto acontece, por exemplo, através do GLUT2 em hepatócitos, nos quais a glicogenólise e/ou a gliconeogênese elevam a concentração intracelular de glicose, ou ainda, através de GLUT 1 ou GLUT 2 em células epiteliais de intestino e túbulo renal, nas quais a glicose é transportada acoplada ao Na+ pelos SGLTs, elevando a concentração intracelular do substrato, para então ocorrer um efluxo a favor de gradiente na membrana basolateral dessas células.

(1)
- Transportadores de glicose (GLUTs) – A insulina exerce seus efeitos biológicos através da ligação a um receptor na superfície da célula alvo. O receptor de insulina é uma glicoproteína que consiste de duas subunidades alfa extracelulares e duas subunidades beta, que são parcialmente intracelulares.
GLUT-1 está envolvido na captação de glicose basal e da glicose não mediada pela insulina em muitas células;
GLUT-2 é importante na célula beta da ilhota, onde é um pré-requisito com a glicocinase, para a percepção da glicose;
GLUT-3 está envolvido na captação não mediada pela insulina da glicose no cérebro;
GLUT-4 é responsável pela captação de glicose estimulada pela insulina nos músculos e tecido adiposo.
Internalização – Após a ligação da insulina a seu receptor, o complexo insulina-receptor é internalizado através da invaginação pela membrana circulante para formar um “endossoma”. Os receptores são reciclados para a superfície celular, mas a insulina e degrada em lisossomas.
(2)
Referência:
NOVA SÍNTESE:








Comentários