
NCS: diabetes, glicosúria, hiperglicemia, peptideo- c
- ReMed

- 10 de jan. de 2021
- 31 min de leitura
1ano 1semestre 3módulo 5SP
27 de Maio de 2019


PROBLEMAS :
Igor não se sentia bem (cansaço, dificuldade para enxergar, formigamento nas pernas, sempre com sede e urinando muito)
Preocupação por ter sido demitido e não conseguir outro emprego
Igor notou anormalidades em sua saúde nos últimos 6 meses
Histórico de síndrome metabólica
Pais diabéticos
Obesidade tipo I
Circunferência alterada
Glicosúria (++)
Albuminúria
Corpos cetônicos na urina
Hiperglicemia
Hemoglobina glicada superior ao normal
Triglicerídeos alto
DM tipo II confirmada
HIPÓTESES :
O DM tipo II está relacionado com a resistência a insulina, já o DM tipo I é uma doença autoimune que destrói as células β
O DI (diabetes insipidus) causada pela deficiência de ADH, hormônio responsável pela regulação da diurese
Igor vai muito ao banheiro pois a glicose concentrada, que esta no sangue, vai para o sistema renal e causa uma maior diferença de gradiente, sendo assim há mais pressão osmótica e portanto mais urina.
Esta perde água constante leva a um quadro de desidratação, o que gera a necessidade de maior ingestão de água
Igor fica mais cansado, tem dislipidemia e traços para corpos cetônicos na urina pois o seu metabolismo energético é deficiente, já que a utilização da glicose é prejudicada pela deficiência de entrada de insulina nas células, aumentando assim a utilização de lipídeos ( lipólise) para a gliconeogênese
O excesso de glicose no sangue ocasiona uma glicorilação, desencadeando um processo inflamatório que afeta principalmente os capilares e arteríolas e consequentemente obstrução delas. Como resposta o corpo realiza o fenômeno de vascularização colateral para suprir a troca gasosa deficiente. Sendo assim, se o fenômeno ocorrer na retina pode ocasionar a perda da visão
Como há hiperglicemia, haverá também guande quantidade de glicose entrando por difusão nas terminações nervosas, o que pode levar a uma lesão no nervo e isso explica o formigamento
Os fatores de riscos são obesidade, histórico familiar, C.AB maior que 94 cm, síndrome metabólica
O processo de glicosilação, pode acarretar lesão dos glomerulos permitindo passagem de moléculas maiores, como a albumina(albuminúria)
O glucagon está suprimido devido a alta taxa de glicose no sangue
QUESTÕES
Explique e diferencie os tipos de diabetes (DM I e II e a insipidus)
Explique a glicosúria relacionada a DM tipo II
Como a DM tipo II afeta o metabolismo de proteínas, carboidratos e lipídeos?
Como a hiperglicemia pode causar cegueira, albuminúria e lesão nervosa?
Quais as alterações hormonais relacionadas a diabetes e o comportamento do Peptideo-C?
Quais são os fatores de risco para DM tipo II?
1. Explique e diferencie os tipos de diabetes (DM I e II e a insipidus)
OBS.:
O diabetes melito não é uma doença única e sim um grupo heterogêneo de síndromes multifatoriais e poligênicas, caracterizadas por elevação da glicemia em jejum, causada por deficiência relativa ou absoluta de insulina. O diabetes melito é a principal causa de cegueira e amputação no adulto e uma importante causa de falha renal, dano neural, ataques cardíacos e acidentes vasculares cerebrais.
Os casos de diabetes melito podem, em sua maioria, ser divididos em dois grupos, tipo 1 (inicialmente denominado diabetes melito dependente de insulina) e tipo 2 (inicialmente denominado diabetes melito independente de insulina).
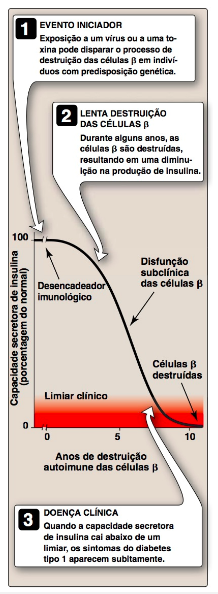
DM TIPO 1 A doença é caracterizada por deficiência absoluta de insulina, causada por ataque autoimune às células β do pâncreas. No diabetes tipo 1, as ilhotas de Langerhans tornam-se infiltradas com linfócitos T ativados, levando a uma condição denominada de insulite. Ao longo de alguns anos, esse ataque autoimune leva à depleção gradual da população de células β. Contudo, os sintomas aparecem abruptamente quando 80 a 90% das células β foram destruídas. Nesse ponto, o pâncreas falha em responder adequadamente à ingestão de glicose, e a terapia com insulina é necessária para restaurar o controle metabólico e prevenir a cetoacidose grave. A destruição das células β requer um estímulo ambiental (como uma infecção viral) e um determinante genético, que permite às células β serem reconhecidas como “estranhas”. OBS.: Pacientes com diabetes tipo 1 praticamente não têm células β e não conseguem responder às variações em combustíveis circulantes, nem manter uma secreção basal de insulina. Pacientes com diabetes tipo 1 geralmente podem ser reconhecidos pelo aparecimento abrupto de poliúria (micção frequente), polidipsia (sede excessiva) e polifagia (fome excessiva), com frequência desencadeados por estresse ou por uma doença. Esses sintomas são geralmente acompanhados por fadiga, perda de peso e fraqueza. O diagnóstico é confirmado por glicemia no jejum maior ou igual a 126 mg/dL, comumente acompanhada por cetoacidose. (Nota: uma glicemia no jejum de 100 a 125 mg/dL é classificada como inadequada.) O jejum é definido como a ausência de ingestão calórica por pelo menos oito horas. Quando o diagnóstico do diabetes tipo 1 é incerto pela apresentação clínica, recomenda-se teste de anticorpos para as células das ilhotas. (Nota: o teste de tolerância à glicose não é rotineiramente usado como ferramenta para diagnóstico de diabetes, pois é difícil de ser realizado na prática e os resultados são altamente variáveis; contudo, é usado para triagem de diabetes gestacional em mulheres grávidas).
ALTERAÇÕES METABÓLICAS DM TIPO 1
As anormalidades metabólicas do diabetes melito do tipo 1 resultam da deficiência de insulina, a qual afeta de forma marcante o metabolismo de três tecidos: fígado, músculo e tecido adiposo.
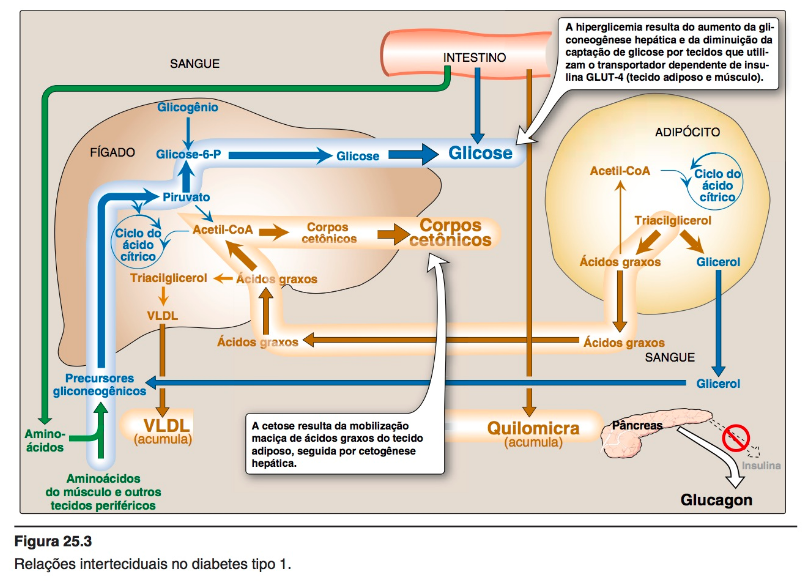
1. Hiperglicemia e cetoacidose
Níveis elevados de glicose e cetonas no sangue são as características do diabetes melito do tipo 1 não tratado.
A hiperglicemia é causada por aumento na produção hepática de glicose, combinada com diminuição na sua utilização periférica (músculo e tecido adiposo têm o GLUT-4 sensível à insulina).
A cetose resulta da mobilização aumentada de ácidos graxos do tecido adiposo, combinada com β-oxidação acelerada de ácidos graxos no fígado e aumento na síntese de 3-hidroxibutirato e acetoacetato.
Nota: a acetil-coA proveniente da β-oxidação é substrato para a cetogênese e efetor alostérico[ o efetor alostérico pode aumentar (efetor positivo) ou diminuir (efetor negativo) a atividade catalítica, através de modificações no sítio catalítico ] da piruvato carboxilase, uma enzima gliconeogênica.
2. Hipertriacilglicerolemia
Nem todos os ácidos graxos que chegam ao fígado podem ser oxidados ou utilizados na síntese de corpos cetônicos.
O excesso de ácidos graxos é convertido em triacilgliceróis, que são empacotados e secretados em lipoproteínas de densidade muito baixa (VLDL).
Os quilomicra são sintetizados a partir dos lipídeos da dieta, pelas células da mucosa intestinal após a refeição. Como a degradação das lipoproteínas catalisada pela lipase lipoproteína nos capilares do músculo e do tecido adiposo é baixa nos diabéticos (a síntese da enzima é diminuída quando os níveis de insulina estão baixos), os níveis plasmáticos de quilomicra e VLDL se elevam, resultando em hipertriacilglicerolemia.
OBS.: Tratamento da DM tipo 1 - Bioquímica Ilustrada, pag. 340 e 341
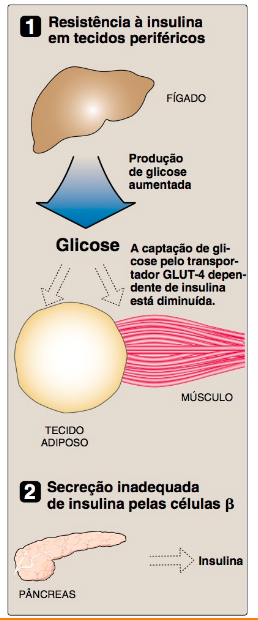
DM TIPO 2 Caracteristicamente, o diabetes tipo 2 se desenvolve de modo gradual, sem sintomas óbvios. A doença é com frequência detectada por exames de triagem de rotina. Entretanto, muitos indivíduos com diabetes tipo 2 apresentam sintomas de poliúria e polidipsia de várias semanas de duração. A polifagia pode estar presente, mas é menos comum. Os pacientes com diabetes tipo 2 apresentam uma combinação de resistência à insulina com disfunção das células β, mas não necessitam de insulina para manter a vida, embora a insulina possa ser necessária em algum momento para controlar a hiperglicemia e manter a HbA1c abaixo de 7%. As alterações metabólicas observadas no diabetes tipo 2 são mais brandas do que as descritas para o tipo 1, em parte, porque a secreção da insulina no diabetes tipo 2 – embora inadequada – impede a cetogênese excessiva e restringe o desenvolvimento da cetoacidose diabética. O diagnóstico baseia-se mais comumente na presença de hiperglicemia – isto é, uma concentração de glicose sanguínea no jejum igual ou superior a 126 mg/dL. A patogênese não envolve viroses ou anticorpos autoimunes. (Nota: uma complicação aguda do tipo 2 na velhice é um estado hiperglicêmico hiperosmolar caracterizado por hiperglicemia e desidratação graves, além de estado mental alterado.) OBS.: O diabetes melito tipo 2 é caracterizado por hiperglicemia, resistência à insulina e relativa diminuição na secreção de insulina.
A RESISTÊNCIA À INSULINA
É a diminuição na capacidade dos tecidos-alvo, como fígado, tecido adiposo e músculo, de responder adequadamente às concentrações circulantes normais (ou elevadas) de insulina.
Por exemplo, a resistência à insulina é caracterizada por produção não controlada de glicose hepática e por captação diminuída de glicose pelo músculo e pelo tecido adiposo.
--Resistência à insulina e diabetes tipo 2
A resistência à insulina sozinha não levará ao diabetes tipo 2.
O diabetes tipo 2 se desenvolve em indivíduos resistentes à insulina que também apresentam diminuição na função das células β.
Em geral, a resistência à insulina com o risco subsequente de desenvolvimento do diabetes tipo 2 é comumente observada em idosos e em indivíduos obesos, sedentários, ou nos 3 a 5% de mulheres grávidas que desenvolvem diabetes gestacional.
Esses pacientes são incapazes de compensar adequadamente a resistência à insulina com a liberação aumentada de insulina.
( curso temporal para o desenvolvimento de hiperglicemia e a perda de células β funcionais )

OBS.: Causas da resistência à insulina.
A resistência à insulina aumenta com o ganho de peso e diminui com a perda de peso. Isso sugere que o acúmulo de lipídeos é importante no desenvolvi- mento da resistência à insulina. O tecido adiposo não é simples- mente um órgão de armazenamento de energia, mas também um órgão secretório. As substâncias reguladoras produzidas pelos adipócitos incluem a leptina e a adiponectina, as quais podem contribuir para o desenvolvimento da resistência à insulina. Além disso, os níveis elevados de ácidos graxos livres que ocorrem na obesidade têm sido também implicados no desenvolvimento da resistência à insulina. (Nota: ácidos graxos livres também provêm da lipólise no tecido adiposo resistente à insulina.)
---Células β disfuncionais
No diabetes tipo 2, a capacidade das células β do pâncreas é inicialmente mantida, resultando em níveis de insulina que variam desde acima até abaixo do normal. Entretanto, com o decorrer do tempo, as células β tornam-se progressivamente disfuncionais, sendo incapazes de secretar insulina suficiente para corrigir a hiperglicemia preponderante.
A progressão natural da doença resulta no declínio da capacidade de controlar a hiperglicemia com secreção endógena de insulina. A deterioração da função das células β pode ser acelerada pelos efeitos tóxicos da hiperglicemia persistente e pelo aumento dos ácidos graxos livres.

ALTERAÇÕES METABÓLICAS NA DM TIPO 2
As anormalidades metabólicas do DM tipo 2 são o resultado da resistência à insulina, que se expressa principalmente no fígado, no músculo e no tecido adiposo.
A. Hiperglicemia
A hiperglicemia é causada por aumento na produção de glicose hepática, combinado com diminuição na sua utilizção periférica.
Em geral, a cetose é mínima ou ausente em pacientes com diabetes tipo 2, pois a presença de insulina – mesmo na presença de resistência à insulina – diminui a cetogênese hepática. (Nota: metformina, um agente oral para o tratamento do diabetes tipo 2, inibe a gliconeogenese hepática.)
B. Dislipedemia
No fígado, os ácidos graxos são convertidos em triacilgliceróis, que são empacotados e secretados como VLDL.
Os quilomicra são sintetizados a partir dos lipídeos da dieta pelas células da mucosa intestinal após uma refeição. Como a degradação das lipoproteínas, catalisada pela lipase lipoproteica no tecido adiposo e no músculo é baixa nos diabéticos, os níveis plasmáticos de quilomicra e de VLDL se elevam, resultando em hipertriacilglicerolemia. Baixos níveis de HDL estão também associados a diabetes tipo 2.



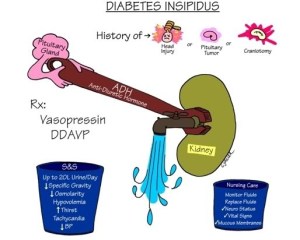
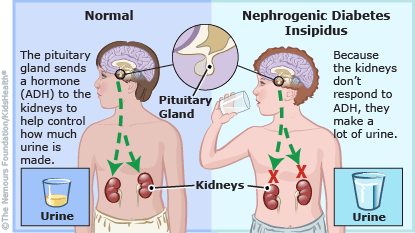
DIABETES INSIPIDUS CENTRAL(DIC) E DIABETES INSIPIDUS NEFROGÊNICA (DIN)
Diabetes insípido resulta de uma deficiência de vasopressina decorrente de doença hipotalâmica-hipofisária (diabetes insípido central [DIC]) ou da resistência dos rins à vasopressina (diabetes insípido nefrogênico [DIN]). Desenvolvem-se poliúria e polidipsia. O diagnóstico se dá pelo teste de privação de água mostrando incapacidade de atingir a concentração máxima de urina; as concentrações de vasopressina e a resposta à vasopressina endógena auxiliam a diferenciar o diabetes insípido central do nefrogênico.
O diabetes insípido nefrogênico é caracterizado pela incapacidade de concentrar a urina em resposta à vasopressina.
O diabetes insípido central é caracterizado pela falta da vasopressina.
Qualquer tipo de diabetes insípido pode ser completo ou parcial.
OBS.: A vasopressina (ADH), também conhecida como arginina vasopressina ou argipressina ou hormônio antidiurético ou hormona antidiurética, é um hormônio humano secretado em casos de desidratação e queda da pressão arterial; fazendo com que os rins conservem a água no corpo, concentrando e reduzindo o volume da urina.
DIC
Fisiopatologia
O lobo posterior da hipófise é o principal local de armazenamento e liberação de vasopressina, mas a vasopressina é sintetizada no hipotálamo.
O hormônio recém-sintetizado ainda pode ser liberado na circulação contanto que os núcleos hipotalâmicos e parte do trato neuro-hipofisário estejam intactos. Apenas cerca de 10% dos neurônios neurossecretores devem permanecer intactos para evitar o DIC.
A patologia do DIC, assim, sempre envolve os núcleos supraóptico e paraventricular do hipotálamo e uma grande porção da haste hipofisária.
O DIC pode ser completo (ausência da vasopressina) ou parcial (quantidades insuficientes devasopressina). O DIC pode ser primário, em que há diminuição significativa nos núcleos hipotalâmicos do sistema neuro-hipofisário.
Etiologia
DIC primário
Anormalidades genéticas do gene da vasopressina no cromossomo 20 são responsáveis pelas formas autossômicas dominantes do DIC primário, mas muitos casos são idiopáticos.
DIC secundário
O DIC também pode ser secundário (adquirido), causado por várias lesões, incluindo hipofisectomia, lesões cranianas (em particular fraturas da base do crânio), tumores supra e intrasselares (primários ou metastáticos), histiocitose de células de Langerhans (doença de Hand-Schüller-Christian), hipofisite linfocítica, granulomas (sarcoidose ou tuberculose), lesões vasculares (aneurisma, trombose) e infecções (encefalite, meningite).
Sinais e sintomas
O início pode ser insidioso ou abrupto, em qualquer idade. Os únicos sintomas no DIC primário são polidipsia e poliúria. No DIC secundário, os sinais e sintomas das lesões associadas também estão presentes. Grandes quantidades de líquidos podem ser ingeridas e grandes volumes (3 a 30 l/dia) de urina muito diluída (gravidade específica, normalmente < 1,005 e osmolalidade < 200 mOsm/l) são excretados. Noctúria quase sempre ocorre. Desidratação e hipovolemia podem se desenvolver rapidamente se as perdas urinárias não forem continuamente repostas.
OBS.: Pontos-chave
Diabetes insípido central (DIC) é causado por uma deficiência de vasopressina, o que diminui a capacidade dos rins de reabsorver água, resultando em poliúria maciça (3 a 30 l/dia).
A causa pode ser uma doença genética primária ou vários tumores, lesões infiltrativas, lesões ou infecções que afetam o sistema hipotalâmico-hipofisário.
Diagnosticar usando um teste de privação de água; os pacientes não podem concentrar urina de maneira máxima após a desidratação, mas podem concentrar urina depois de receber vasopressina exógena.
Níveis baixos de vasopressina são diagnósticos, mas os níveis de vasopressina são difíceis de medir e o teste não está rotineiramente disponível.
Abordar quaisquer causas tratáveis e dar desmopressina, um análogo sintético davasopressina.
DIN
Diabetes insípido nefrogênico (DIN) é a incapacidade de concentrar a urina devido à resposta renal tubular prejudica à vasopressina (ADH), que provoca excreção de grandes quantidades de urina diluída. Pode ser herdado ou secundário a condições que prejudiquem a capacidade de concentração renal. Os sinais e sintomas incluem poliúria e aqueles relacionados com desidratação e hipernatremia ( hipernatremia é a concentração sérica de sódio > 145 mEq/L. Implica déficit de ACT em relação ao sódio corporal total porque a ingestão de água é inferior às perdas)
Etiologia
DIN herdado
DIN de herança mais comum é um traço ligado ao X com penetrância variável em mulheres heterozigotas que afeta o gene do receptor 2 de arginina vasopressina (AVP). Mulheres heterozigotas podem não ter sintomas ou ter um grau variável de poliúria e polidipsia, ou podem ser tão gravemente afetadas quanto os homens.
Em casos raros, o DIN é causado por uma mutação autossômica recessiva ou autossômica dominante que afeta o gene aquaporina-2 e pode afetar tanto homens como mulheres.
DIN adquirido
O DIN adquirido pode ocorrer quando doenças (muitas das quais são doenças tubulointersticiais) ou fármacos alteram a camada medular ou o néfron distal e impedem a capacidade de concentrar urina, tornando os rins aparentemente insensíveis à vasopressina. Essas doenças incluem:
Doença do rim policístico autossômico dominante
Complexo da doença renal cística medular e nefronoftise renal
Nefropatia da anemia falciforme
Liberação de fibrose periureteral restritiva
Rim espongiomedular
Pielonefrite
Hipercalcemia
Amiloidose
Síndrome de Sjögren
Síndrome de Bardet-BiedL
Certas neoplasias (p. ex., mieloma, sarcoma)
Diversos medicamentos, especialmente lítio, mas também outros (p. ex., demeclociclina, anfotericina B, dexametasona, dopamina, ifosfamida, ofloxacino, orlistate)
Possivelmente nefropatia hipocalêmica crônica
O DIN pode também ser idiopático. Uma forma leve de DIN adquirido pode ocorrer em qualquer paciente idoso ou paciente que apresente insuficiência renal aguda ou crônica.
Ainda, certas síndromes clínicas podem assemelhar-se ao DIN:
A placenta pode secretar vasopressina durante a segunda metade da gestação (síndrome chamada diabetes insípido gestacional).
Após cirurgia hipofisária, alguns pacientes secretam um precursor de ADH ineficaz ao invés de vasopressina.
Sinais e sintomas
A geração de grandes quantidades de urina diluída (3 a 20 l/dia) é característica. Tipicamente, os pacientes apresentam boa resposta à sede e o sódio sérico permanece praticamente normal. Entretanto, pacientes que não têm bom acesso à água ou que não conseguem manifestar sua sede (p. ex., bebês e pacientes idosos com demência) geralmente desenvolvem hipernatremia devido à desidratação extrema. A hipernatremia pode causar sintomas neurológicos, tais como excitabilidade neuromuscular, confusão, convulsões ou coma.
OBS.: Pontos-chave
Pacientes com DIN não conseguem concentrar a urina devido a resposta tubular renal prejudicada à vasopressina.
Eles normalmente eliminam grandes volumes de urina diluída, têm adequadamente sede e apresentam níveis séricos de sódio quase normais.
Minimizar as sequelas neurológicas evitáveis considerando DIN herdado em crianças com poliúria ou familiares afetados.
Medir o volume de urina de 24 h, a osmolalidade e eletrólitos séricos.
Confirmar o diagnóstico com um teste de privação de água.
Assegurar ingestão adequada de água livre, restringir sal e proteína na dieta e usar um diurético tiazídico ou amilorida, conforme necessário.
(2)


NOVA SÍNTESE:
INSIPIDUS
-Nefrogênica - falha nos receptores de ADH. Manifestação geralmente precoce. Pode ser herdada ou adiquirida
-Central - falha na produção de ADH
OBS.:mecanismo de ação de ADH (vasopressina): age principalmente nos ductos contorcidos distal e coletor, abrindo os "canais de água" ou "aquaporinas" favorecendo a reabsorção de água
MELLITUS
--DM1
Parada na secreção de insulina, causada pela destruição das células beta do pâncreas ( autoimune), indivíduos geneticamente propensos, manifestação geralmente precoce, pode se manifestar tardiamente (rara), 80-90%
--DM2
Relaciona-se à obesidade ( reação inflamatória ---> inibição do GLUT4)
Resistência à insulina:
Células beta são afetadas pela hiperglicemia e lipotoxidade --- baixa de insulina endógena
REFERÊNCIAS:
(1) Livro: Bioquímica Ilustrada - Harvey e Ferrier
(2) Manual MSD -
Ian M. Chapman, MBBS, PhD, Professor of Medicine, Discipline of Medicine, University of Adelaide, Royal Adelaide Hospital -https://www.msdmanuals.com/pt-br/profissional/dist%C3%BArbios-end%C3%B3crinos-e-metab%C3%B3licos/dist%C3%BArbios-hipofis%C3%A1rios/diabetes-ins%C3%ADpido-central
James I. McMillan, MD, Associate Professor of Medicine, Nephrology Fellowship Program, Loma Linda University - https://www.msdmanuals.com/pt-br/profissional/dist%C3%BArbios-geniturin%C3%A1rios/anormalidades-do-transporte-renal/diabetes-ins%C3%ADpido-nefrog%C3%AAnico
Visão geral das doenças tubulointersticiais - Navin Jaipaul, MD, MHS, Associate Professor of Medicine, Loma Linda University School of Medicine; Chief, Nephrology, VA Loma Linda Healthcare System - https://www.msdmanuals.com/pt-br/profissional/dist%C3%BArbios-geniturin%C3%A1rios/doen%C3%A7as-tubulointersticiais/vis%C3%A3o-geral-das-doen%C3%A7as-tubulointersticiais
2. Explique a glicosúria relacionada a DM tipo II
Glicosúria renal é a presença de glicose na urina, sem hiperglicemia; resulta de um defeito adquirido ou hereditário isolado do transporte de glicose ou ocorre em outras doenças tubulares renais.
Glicosúria renal é a excreção de glicose na urina, com níveis plasmáticos normais de glicose.
A glicosúria renal pode ser herdada. Essa forma geralmente envolve uma redução do transporte máximo de glicose (a taxa máxima na qual a glicose pode ser reabsorvida) e perda subsequente de glicose na urina. A doença herdada é geralmente transmitida como traço recessivo incompleto (heterozigotos têm glicosúria leve).
A glicosúria renal pode ocorrer sem qualquer outra anormalidade na função renal ou como parte de defeito generalizado da função do túbulo proximal (síndrome de Fanconi). Também pode ocorrer com várias doenças sistêmicas, incluindo cistinose, doença de Wilson, tirosinemia hereditária e síndrome oculocerebrorrenal (síndrome de Lowe)
(1)
A glicosúria renal familiar é um distúrbio tubular raro, caracterizado por glicosúria isolada persistente na ausência de hiperglicemia ou qualquer alteração do metabolismo da glicose, assim como de outro sinal de disfunção tubular generalizada.
Está associada a mutações do gene SLC5A2, localizado no cromossoma 16 e responsável pela codificação do SGLT2, um cotransportador sódio/glicose de baixa afinidade localizado na membrana luminal do túbulo renal proximal e que efetua a reabsorção tubular da grande maioria da glicose filtrada.
É de transmissão autossômica recessiva. Apesar de o grau de glicosúria poder ser variável, os indivíduos atingidos são geralmente assintomáticos e o prognóstico é favorável, sendo muito rara a ocorrência de qualquer tipo de complicação renal ou extrarrenal.
Os casos apresentados confirmam o descrito na literatura, em que mutações do gene SLC5A2 são causadoras de glicosúria renal, e que esta tem uma transmissão autossômica recessiva.
Tal como descrito previamente em relação a outros doentes com glicosúria renal causada por mutações do gene SLC5A2, estas adolescentes apresentavam glicosúria renal significativa, na ausência de hiperglicemia, acidose metabólica, aminoacidúria, proteinúria ou fosfatúria.
Não apresentavam, também, qualquer tipo de complicação renal ou extrarrenal, tal como o verificado na grande maioria dos casos.
Alguns autores têm levantado a hipótese de um eventual efeito benéfico a longo prazo da inibição do SGLUT2, tal como a causada pelas mutações do gene SLC5A2, e que um inibidor específico deste cotransportador poderá eventualmente ser um possível alvo terapêutico para o controlo da obesidade, Diabetes Mellitus e até hipertensão.
(2)
A hiperglicemia é causada por aumento na produção de glicose hepática, combinado com diminuição na sua utilização periférica.
A progressão natural da doença resulta no declínio da capacidade de controlar a hiperglicemia com secreção endógena de insulina. A deterioração da função das células β pode ser acelerada pelos efeitos tóxicos da hiperglicemia persistente e pelo aumento dos ácidos graxos livres.
(Livro: Bioquímica ilustrada)
Se a [glicose] no sangue for suficientemente alta, os rins podem não reabsorver toda a glicose filtrada, então aparece na urina (glicosúria).
Quando um excesso de glicose é excretado na urina, ele é acompanhado por perda excessiva de líquidos e eletrólitos – diurese osmótica. Como resultado do excesso da perda de líquido, o paciente sofre um aumento do volume da urina (poliúria) e aumento da sede (polidpsia).
(3)
NOVA SÍNTESE:
Glicose atravessa facilmente os glomérulos durante a filtração glomerular
Saturação da reabsorção da glicose nos ductos contorcidos --> glicosúria
Glicemia maior que 180 mg/dL ----glicosúria
REFERÊNCIAS
(1) Glicosúria renal - Por James I. McMillan, MD, Associate Professor of Medicine, Nephrology Fellowship Program, Loma Linda University - https://www.msdmanuals.com/pt-br/profissional/dist%C3%BArbios-geniturin%C3%A1rios/anormalidades-do-transporte-renal/glicos%C3%BAria-renal
(2) Joana Rabelo - Glicosúria renal: a propósito de dois casos clínicos - 2012 - http://www.scielo.br/pdf/jbn/v34n3/v34n3a13.pdf
(3) Paula Nogueira, UNIVERSIDADE DE SÃO PAULO Escola de Enfermagem Departamento de Enfermagem Médico-Cirúrgica, Emergências Diabéticas - https://edisciplinas.usp.br/pluginfile.php/4136772/mod_resource/content/2/emergencias%20diab%C3%A9ticas%202017.pdf
3. Como a DM tipo II afeta o metabolismo de proteínas, carboidratos e lipídeos?
De uma maneira geral, as vias metabólicas mais afetadas nesta patologia, embora variem de acordo com o tipo da diabetes, são as dos hidratos de carbono e as dos lípidos, especialmente porque ambas são essenciais para o fornecimento e armazenamento de energia. O metabolismo das proteínas é também alterado, a nível da síntese proteica, com alterações na transcrição genética de vários intervenientes nas vias metabólicas, e da produção de intermediários para o ciclo de Krebs e de proteínas intracelulares.
Na diabetes, embora o indivíduo tenha hiperglicemia, como os tecidos não conseguem captar a glucose devido a falhas nos transportadores da mesma, o organismo reage como se houvesse falta desta hexose, aumentando as vias para a sua produção e para obtenção de energia, através do aumento da gliconeogénese, da lipólise e da produção de corpos cetónicos.
A diabetes mellitus está diretamente relacionada com problemas nas células β e com a insulina.
Na DM1 há uma redução da massa de células β devido à destruição autoimune das mesmas, levando a uma deficiência total de insulina que pode progredir para estados de hiperglicemia e cetoacidose graves. No entanto, as restantes células das ilhotas de Langerhans não são afetadas, havendo uma produção excessiva de glucagon, o que contribui também para o estado hiperglicémico do diabético.
Na DM2 está envolvida uma forte vertente genética que torna os indivíduos predispostos à obesidade e à resistência à insulina, sendo essas condições agravadas por um estilo de vida com pouca atividade física e má alimentação.
Apesar disso, apenas quando as células β deixam de conseguir compensar a resistência à insulina é que surge a diabetes. Por isso, a maior parte das pessoas que tem resistência à insulina não chega a desenvolver a doença. Anomalias na produção de glucagon são também verificadas na diabetes tipo 1 e tipo 2, não sendo a sua secreção suprimida quando o organismo está em estado de hiperglicemia ou estimulada quando se encontra em hipoglicemia.
DM1
Na diabetes tipo 1 há uma completa incapacidade de produção de insulina pelas células β do pâncreas e destruição das mesmas. Assim, a relação insulina/glucagon não aumenta, mantendo o fígado em constante gluconeogénese e cetogénese, não sendo os níveis de glucose no sangue devidamente controlados. No estado pós-prandial este efeito é aumentado, pois a gluconeogénese contínua produz mais glucose, para além da obtida pela dieta, contribuindo assim para a hiperglicemia. No tecido muscular e adiposo, os GLUT-4 permanecem no interior das células, não permitindo assim o transporte da glucose (Im, S. et alii., 2007). Juntamente com a gluconeogénese alterada no fígado, ocorre também a degradação de proteínas de forma descontrolada no músculo esquelético, permitindo manter a hiperglicemia mesmo em jejum.
A degradação de lípidos pelo tecido adiposo também fica desregulada, aumentando a concentração plasmática de ácidos gordos e a produção de corpos cetónicos pelo fígado. Deste modo, uma das primeiras e principais manifestações da doença, a cetoacidose, desenvolve-se devido à acumulação dos corpos cetónicos e de H+ . A oxidação dos ácidos gordos e a formação de corpos cetónicos não conseguem eliminar completamente os ácidos gordos produzidos no fígado, sendo o excesso esterificado em triacilgliceróis e direcionados para a síntese de VLDL. Tanto as VLDL como os quilomicrons não podem ser eliminados da corrente sanguínea pela proteína lipase, uma vez que a sua síntese depende da estimulação pela insulina, resultando assim em hipertriacilglicerolemia e hiperquilomicronemia.

DM2
A diabetes tipo 2 é uma patologia na qual há produção de insulina, embora haja disfunção das células β, com diminuição da secreção da hormona e também resistência à mesma, que não consegue ser compensado por causa da sua produção insuficiente. No início da doença, as células β ainda conseguem segregar insulina suficiente para compensar a resistência à mesma, mantendo durante algum tempo os níveis de glucose normais. No entanto, o declínio progressivo das células β justifica a dificuldade que as pessoas têm de controlar a hiperglicemia ao longo do tempo e a necessidade do aumento progressivo do número e doses dos agentes antidiabéticos per os, bem como a eventual necessidade de insulina exógena devido à resistência ao tratamento oral.
Esta condição está fortemente relacionada com a obesidade. Pessoas diabéticas obesas têm níveis elevados de ácidos gordos livres e de insulina, bem como pessoas obesas não diabéticas que normalmente também têm níveis de insulina superiores aos de um indivíduo com peso normal. Para além disso, o fator de necrose tumoral α (TNFα) e a resistina, produzidos pelos adipócitos, estão presentes em grande quantidade, o que faz com que os valores destas moléculas, que têm efeito oposto ao da insulina, sejam também elevados. Além disso, a acumulação de lípidos intramiocelulares interfere com o recrutamento dos GLUT-4 para a superfície celular, resultando assim na diminuição da taxa do transportador da glucose estimulado pela insulina para as células musculares.
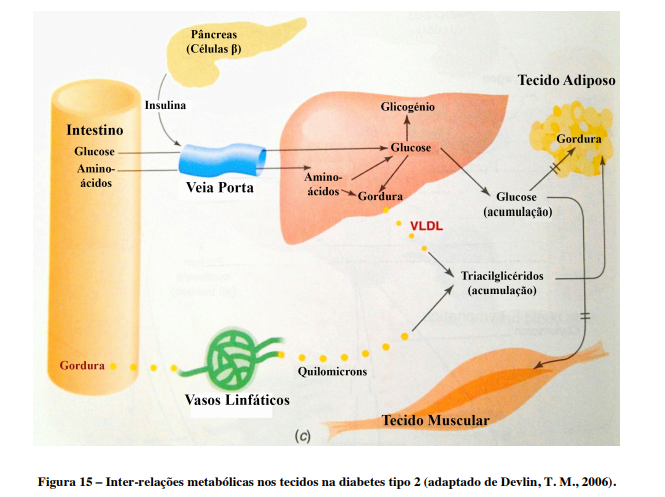
A hiperglicemia acontece pela insuficiência de insulina para controlar a produção de glucose no fígado e para permitir a captação desta pelo músculo esquelético. Assim, não ocorre o aumento da frutose-2,6-bisfosfatase nem a regulação negativa do fosfoenolpiruvato carboxilase, que aconteceria normalmente. No tecido adiposo e no músculo esquelético, em resposta à estimulação pela insulina, há diminuição da translocação das vesículas intracelulares de GLUT-4 para a membrana.
Ao contrário do que acontece na diabetes tipo 1, a cetoacidose não costuma manifestar-se devido ao facto de haver produção suficiente de insulina para prevenir a libertação descontrolada de ácidos gordos a partir dos adipócitos. Além disso, os ácidos gordos que chegam ao fígado, após degradação, ou são sintetizados de novo ou são direcionados para a formação de triacilgliceróis.
A hipertriacilglicerolemia é característica da doença e a hiperquilomicronemia não se costuma desenvolver. Isto deve-se ao aumento da síntese hepática dos ácidos gordos e ao desvio dos mesmos do fígado para formar triacilglicerol e VLDL, com níveis elevados também destas lipoproteínas. Normalmente, a degradação de lípideos e a gluconeogénese não podem ocorrer em simultâneo, mas no caso da diabetes tipo 2, isto acontece devido à alteração, quando há resistência à insulina, da via sinalizadora da mesma que controla estes processos.
Um defeito na via de sinalização da insulina no controlo da gluconeogénese evita a supressão da produção hepática de glucose, via PI3 quinase, com níveis elevados de insulina.
Em relação ao controlo da síntese dos ácidos gordos e da sua esterificação, a via de sinalização da insulina tem uma maior capacidade de resposta, o que leva à produção excessiva de triacilglicerol.
O metabolismo das proteínas na diabetes tipo 2 é dos menos compreendidos, embora as vias relativas às proteínas e aos aminoácidos sejam muito influenciadas pela ação da insulina. A complexidade do conhecimento total do efeito desta patologia sobre o metabolismo é demonstrada pelos testes in vitro e in vivo, em que apenas os primeiros demonstraram o efeito anabólico da insulina com diminuição da degradação de proteínas no músculo e aumento da síntese proteica.
(1)
NOVA SÍNTESE:
Livro Fisiologia básica- Rui Curi
Reistência à insulina
-baixo uso da glicose
-Alta gliconeogênese
1º lipídeos (lipólise)-----> produção corpos cetônicos, perda de tecido adiposo
2º proteínas (proteólise)----> produção de corpos cetônicos, perda tônus muscular
Falta de insulina --> não há atuação das enzimas marca-passo do metabolismo da glicose (gliconeogênese)
REFERÊNCIAS:
(1) Nelma Sofia Magro Fernandes, Alterações metabólicas no diabético, Universidade Fernando Pessoa Faculdade de Ciências da Saúde Porto, 2013 - https://bdigital.ufp.pt/bitstream/10284/4476/1/PPG_22125.pdf
4. Como a hiperglicemia pode causar cegueira, albuminúria e lesão nervosa?
Durante o estado hiperglicêmico de longa duração no DM, a glicose forma pontes covalentes com as proteínas plasmáticas através de um processo não enzimático, conhecido como glicação. A glicação proteica e a formação de produtos finais de glicação avançada (AGEs) desempenham um papel importante na patogênese das complicações diabéticas (retinopatia, nefropatia e neuropatia). A glicação das proteínas interfere nas funções normais pela modificação das conformações moleculares, alterando a atividade enzimática e interferindo no funcionamento dos receptores. Os AGEs ligam-se não apenas às proteínas, mas também aos lipídios e ácidos nucleicos, favorecendo as complicações diabéticas.
(2)
A retinopatia diabética proliferativa é marcada pela presença de proliferação neovascular que é um fator importante na patogênese de várias complicações que levam à cegueira, como o descolamento tracional da retina e hemorragia vítrea. Os novos vasos sanguíneos crescem dentro e sobre a retina e na face recentemente exposta pelo descolamento do vítreo posterior. O estímulo para a neovascularização vítreoretiniana é sabidamente devido à liberação de um fator angiogênico produzido pela retina isquêmica. Estudos recentes revelaram que um fator angiogênico extremamente importante é o VEGF (“vascular endothelial growth factor” ou fator de crescimento vascular endotelial).
A síntese do VEGF é regulada pelos níveis de oxigênio no microambiente celular. O estímulo da síntese do VEGF pela hipóxia faz desta substância um possível fator responsável pela retinopatia proliferativa. O VEGF tem sido encontrado no fluido intra-ocular de pacientes com neovascularização ativa da retina e segmento anterior associado a várias doenças oculares associadas à isquemia retiniana. As concentrações de VEGF no vítreo e humor aquoso de pacientes com neovascularização ativa são mais altas que o necessário para estimular a proliferação endotelial “in vitro” e “in vivo”. Ao contrário do que acontece com indivíduos com neovascularização quiescente onde as concentrações do VEGF são muito baixas ou indetectáveis, a panfotocoagulação com “laser” bem sucedida produz uma queda significativa das concentrações do VEGF.
Outros fatores de crescimento como fator básico de crescimento de fibroblastos (bFGF) podem ter um papel na neovascularização retiniana. Apesar de não ser sintetizado diretamente pela retina, este fator está presente no citoplasma das células retinianas e no tecido retiniano onde se liga à matriz extracelular. A lesão celular induzida pela isquemia prolongada pode levar à liberação desse fator.
Esses fatores angiogênicos exercem seu efeito estimulatório nas células que compõem o lado venoso do leito capilar retiniano pré-existente. Com a proliferação das células estimuladas, ocorre a produção de colagenases e outras enzimas digestivas que dissolvem a membrana basal que envolve os capilares. Essas células migram, escorregando pelos espaços na parede dos vasos, em direção ao estímulo angiogênico. A neovascularização se inicia na retina onde é clinicamente evidenciada pela presença das IRMAs (anormalidades microvasculares intra-retinianas). Os novos vasos invadem a MLI e crescem sobre a superfície interna da retina. Apesar de a neovascularização ser incapaz de invadir o vítreo, ela pode crescer na face posterior do vítreo descolado.
A neovascularização a partir do nervo óptico estende-se tipicamente até as paredes do espaço cônico conhecido como área de Martegiani que forma a parte posterior do Canal de Cloquet. Neovascularização vítreorretiniana estimula fibroplasia e fibrose vítrea que progride com o aumento do descolamento do vítreo posterior pela contração das membranas vítreas rasgando os delicados vasos sangüíneos. O resultado disso são hemorragias pré-retinianas e vítreas cuja organização produz ainda mais fibrose e tração. Considerando que o vítreo geralmente permanece aderente às grandes arcadas vasculares temporais, um descolamento localizado da mácula ocorre a partir da contração dessa ponte fibrovascular e tração resultante desta. Tração ântero-posterior também contribui para descolamento de retina.
Olhos com retinopatia diabética proliferativa também são susceptíveis a neovascularização de íris e glaucoma neovascular. Estudos realizados sobre a concentração do VEGF no vítreo e aquoso de olhos de pacientes diabéticos revelaram que os níveis desse fator estavam sempre mais altos no vítreo. Tal fato explica porque a neovascularização de íris se inicia a partir da borda pupilar e ângulo onde a difusão do VEGF a partir do vítreo seria maior. Os neovasos proliferam sobre a superfície normal e avascular da íris aplanando e achatando sua arquitetura normal. Ocorre a formação de aderências permanentes entre a periferia da íris e o trabeculado, chamadas sinéquias anteriores periféricas. Miofibroblastos se proliferam, acompanhando os neovasos e consequentemente fornecendo os motivos para o fechamento angular e o desenvolvimento do ectrópio do esfíncter e do epitélio pigmentar da íris. Alguns pacientes com diabetes apresentam uma forma mais branda de iridopatia, chamada vacuolização rendilhada do epitélio pigmentar da íris. Essa vacuolização rendilhada é causada pelo acúmulo de glicogênio nos espaços cistóides do epitélio pigmentar da íris e ocorre somente em diabéticos, o que sugere sua relação com altos níveis de glicose sérica. A glicogênese focal do epitélio pigmentar da íris deve ser análoga ao acúmulo de glicogênio no túbulos renais conhecida como nefropatia glicogênica de ArmanniEbstein. O dano tecidual causado pela vacuolização rendilhada deve ser responsável pela dispersão pigmentar do humor aquoso durante o intra-operatório de alguns olhos diabéticos. Essa vacuolização rendilhada pode ser evidenciada clinicamente como um padrão “roído de traça” na transiluminação ou biomicroscopia da íris.
Um espessamento da membrana basal ocorre de forma sistêmica no diabetes mellitus conforme explicado anteriormente sobre a retina. Esse espessamento, na camada pigmentada interna do epitélio pigmentar da íris, permite distinguir histopatologicamente se um olho com glaucoma neovascular tem diabetes. O espessamento da membrana basal do epitélio corneano pode predispor a uma descamação desse epitélio durante cirurgia vítreo-retiniana.
(1)
A nefropatia diabética (ND) é uma complicação crônica do diabetes que afeta 20% a 30% das pessoas com diabetes mellitus tipo 1 (DM1) ou DM2 , sendo responsável por aproximadamente metade dos novos casos de insuficiência renal nos indivíduos em diálise e tendo sido associada a aumento significativo da mortalidade, principalmente cardiovascular .
A presença de pequenas quantidades de albumina na urina representa o estágio inicial da nefropatia diabética (microalbuminúria ou nefropatia incipiente). O estágio avançado caracteriza a nefropatia clínica (macroalbuminúria ou proteinúria) e a fase terminal é a insuficiência renal. Indivíduos com ND apresentam outras condições crônicas associadas, como retinopatia diabética, doença macrovascular e hipertensão arterial sistêmica.
O comprometimento glomerular no DM inicia-se, geralmente, cinco a 10 anos depois da evolução do diabetes, apresentando um aumento de incidência após 15 anos de doença. Essa fase inicial geralmente é assintomática.
A nefropatia diabética era definida classicamente pela presença de proteinúria > 0,5g/24h. Nos anos 80, entretanto, estudos demonstraram que pequenas quantidades de albumina na urina, geralmente não detectadas pelos métodos convencionais, eram preditivas de proteinúria no DM1 e DM2.
A partir daí a ND classifica-se em estágios baseados na excreção urinária de albumina (EUA):
microalbuminúria ou fase de nefropatia incipiente com EUA entre 30-300mg/24h ou 20-200mg/min;
macroalbuminúria ou fase de nefropatia clínica, caracterizada por EUA superior a 300mg/24h ou 200mg/min; e fase de insuficiência renal terminal.
Acredita-se que haja um fator genético predisponente, além dos fatores de risco habituais, como hiperglicemia, hipertensão arterial, dislipidemia e tabagismo, que contribuem para a ND. Em estudos epidemiológicos com irmãos com DM1 e DM2 foi evidenciado aumento de aproximadamente três a quatro vezes no outro irmão, quando um deles apresentava ND.
Inicialmente, o paciente apresenta hiperfiltração, com valores elevados na taxa de filtração glomerular (TFG), e eventualmente já pode apresentar microalbuminúria. Essas alterações duram em torno de cinco anos. Durante 20 anos de evolução do diabetes, o indivíduo pode exibir declínio gradual da TFG e microabuminúria persistente, que evolui para proteinúria. A etapa final da história natural da doença caracteriza-se por proteinúria grave, com ou sem síndrome nefrótica e insuficiência renal crônica, levando à fase renal terminal (end stage renal disease — ESRD).
A evolução gradual desses aspectos é causada por alterações estruturais em nível renal, inicialmente evidenciadas por acúmulo gradual e progressivo da matriz extracelular na membrana basal e mesângio glomerular. Mais tarde, a formação de nódulos mesangiais representa a lesão característica da nefropatia de Kimmestiel-Wilson, com extensas lesões adicionais túbulo-intersticiais. A ND não se desenvolve na ausência de hiperglicemia, mesmo que tenha predisposição genética. No entanto, essa predisposição genética contribui para o seu desenvolvimento e apoia a hipótese de que múltiplos fatores estão envolvidos na patogênese da doença
A neuropatia diabética (NeD) é a complicação tardia mais frequente do diabetes e pode ser evidenciada no DM2, muitas vezes no momento do diagnóstico, enquanto no DM1 geralmente aparece cinco anos ou mais após o diagnóstico. Uma definição simples é a presença de sintomas e/ou sinais de disfunção dos nervos periféricos em diabéticos, após a exclusão de outras causas .
Das complicações crônicas microvasculares, o comprometimento do sistema nervoso periférico é uma das manifestações clínicas mais frequentes, afetando entre 40% e 50% dos indivíduos com DM2 e em menor frequência no DM1. Estudos mais recentes sugerem que a NeD também ocorre quando há tolerância diminuída à glicose.
A neuropatia diabética abrange um grupo de alterações relacionadas ao envolvimento estrutural e funcional de fibras nervosas sensitivas, motoras e autonômicas, que podem ser reversíveis ou permanentes. Clinicamente, manifestam-se de formas muito variáveis, desde síndromes dolorosas graves, agudas, secundárias a oscilações glicêmicas, até formas assintomáticas.
A polineuropatia sensitivo-motora difusa simétrica periférica é a mais comum; pode ser de fibras finas, grossas ou mistas. Os sinais e sintomas são parestesia, dor (queimação, pontada, choque ou agulhada) em pernas e pés, hiperestesia (dor ao toque de lençóis e cobertores), diminuição ou perda da sensibilidade tátil (fibras grossas), térmica ou dolorosa (fibras finas), perda dos reflexos tendinosos profundos, fraqueza e perda da motricidade distal, úlceras nos pés.
A neuropatia autonômica caracteriza- -se por função pupilar anormal (diminuição do diâmetro pupilar no escuro), disfunção sudomotora (anidrose, intolerância ao calor, boca seca, sudorese gustatória), disfunção geniturinária (bexiga neurogênica, disfunção erétil, ejaculação retrógrada, disfunção sexual feminina), disfunção gastrintestinal (atonia de vesícula biliar, dismotilidade esofagiana, gastroparesia, constipação, diarreia e incontinência fecal), disfunção cardiovascular (taquicardia em repouso, intolerância a exercícios, hipotensão ortostática, isquemia miocárdica silenciosa) e disfunção metabólica (hipoglicemia despercebida).
Na mononeuropatia focal, os nervos mais comumente acometidos são: ulnar, radial, mediano, femoral e peroneiro. Na neuropatia craniana os nervos mais comumente acometidos são os pares cranianos III, IV, VI e VII.
A patogênese da NeD ainda não está totalmente compreendida, e diferentes teorias tentam explicar a etiologia da doença.
Um dos mecanismos mais estudados abrange a insuficiência microvascular com redução no fluxo sanguíneo neural. Ela é resultante da isquemia absoluta ou relativa dos vasos endoneurais e epineurais, que leva ao espessamento da membrana basal, à diminuição do fluxo sanguíneo e a alterações da permeabilidade vascular. Isso acarreta disponibilidade reduzida de óxido nítrico e excesso de formação de espécies reativas de oxigênio, que favorecem a produção de peroxinitrito, um potente agente oxidante pró-inflamatório e tóxico às células endoteliais. Como consequência disso, há dano à perfusão microvascular, fluxo insuficiente e, consequentemente, lesão neural.
A polineuropatia diabética em geral é diagnosticada tardiamente, e por isso daremos destaque ao tratamento da dor neuropática.
(2)
O controle inadequado da glicose, nível elevado de triglicérides, excesso de peso, tabagismo, pressão alta, o tempo em que você convive com o diabetes e a presença de retinopatia e doença renal (lembra-se delas?) são fatores que favorecem a progressão da neuropatia. Tanto as alterações nos vasos sanguíneos quanto as alterações no metabolismo podem causar danos aos nervos periféricos.
A glicemia alta reduz a capacidade de eliminar radicais livres e compromete o metabolismo de várias células, principalmente dos neurônios. Os principais sinais são:
Dor contínua e constante;
Sensação de queimadura e ardência;
Formigamento;
Dor espontânea que surge de repente, sem uma causa aparente;
Dor excessiva diante de um estímulo pequeno, por exemplo, uma picada de alfinete;
Dor causada por toques que normalmente não seriam dolorosos, como encostar no braço de alguém.
Ao mesmo tempo, em uma segunda etapa dessa complicação, pode haver redução da sensibilidade protetora, como falamos na seção 'Pés e membros inferiores'. As dores, que antes eram intensas demais mesmo com pouco estímulo, passam a ser menores do que deveriam. Daí o risco de haver uma queimadura e você não perceber em tempo.
É comum também que o suor diminua e a pele fique mais seca. O diagnóstico da neuropatia pode ser feito por exames específicos e muito simples nos pés.
(3)
NOVA SÍNTESE:
-Livro: Junqueira - capitulo aparelho urinário
--Cegueira - Retinopatia diabética
-não proliferativa - obstrução dos vasos sanguíneos (isquemia)
-proliferativa - vasodilatação e neovasos, líquidos extravasam, causando cegueira
--Rins- Nefropatia diabética
-esclerose ( engrossamento) dos néfrons
-lesão vascular nos néfrons--> passagem de albumina
--Lesão nervosa - Nefropatia diabética
-Isquêmica: desmielação dos neurônios
-Sobrecarga: aumento de glicose nos néfrons-->disfunção
REFERÊNCIAS:
(1) Zélia Corrêa, Aspectos patológicos da retinopatia diabética, Arq Bras Oftalmol. 2005, http://www.scielo.br/pdf/abo/v68n3/24752.pdf
(2)Balduino Tschiedel [ Médico endocrinologista. Mestre em Genética pela UFRGS. Presidente do Departamento de Diabetes da Sociedade Brasileira de Endocrinologia (SBEM) ] - Complicações crônicas do diabetes- SETEMBRO/OUTUBRO, 2014- http://files.bvs.br/upload/S/0047-2077/2014/v102n5/a4502.pdf
(3) Sociedade Brasileira de Diabetes, Neuropatia Diabética - https://www.diabetes.org.br/publico/complicacoes/neuropatia-diabetica
5. Quais as alterações hormonais relacionadas a diabetes e o comportamento do Peptideo-C?
Hormônios que afetam os níveis de açúcar no sangue
Para entender melhor como hormonas afetam a diabetes, é importante identificar quais as hormonas que podem afetar os níveis de açúcar no sangue.
Insulina
A insulina é um hormônio que é liberado a partir de células beta no pâncreas, e permite ao corpo a usar a glicose para obter energia. A insulina é importante para manter os níveis de açúcar no sangue fique muito baixo ou muito alto. Depois de comer uma refeição e o nível de seu sangue ascensões do açúcar, as células beta são sinalizadas para liberar a insulina no sangue.
Então, que atribui às células para ajudar com a absorção de açúcar do sangue. A insulina é muitas vezes referida como uma “chave” que emitido para desbloquear as células para permitir a conversão de açúcar de modo que pode ser usado como energia.
O glucagon
Produzido pelas alfa dos ilhéus ou células do pâncreas, o glucagon é utilizado para controlar a produção de glicose e cetona no fígado. Esse hormônio é liberado entre as refeições e durante a noite, e é essencial na manutenção do equilíbrio de combustível e os níveis de açúcar no corpo. O glucagon vai sinalizar o fígado quando é hora de quebrar glicogênio e amido , e que também irá ajudar a formar novas unidades de cetonas e glicose a partir de outras substâncias.
Amylin
Amylin é um hormônio que é liberado a partir de células beta juntamente com insulina. Esta hormona atua diminuindo os níveis de glucagon do corpo, que, em seguida, diminui a produção de glicose no fígado e atrasa a taxa de alimentação que se esvazia do estômago. Isso fará com que seu cérebro se sentir como se ele está cheio e satisfeito após uma refeição. O efeito global da amilina é o de reduzir a produção de açúcar pelo fígado durante as refeições, de modo que impede que os níveis de glicose no sangue fiquem muito altos.
Epinefrina
Também é conhecida como a adrenalina, e ele é liberado pelas glândulas supra-renais e terminações nervosas para estimular o fígado a produzir açúcar. A adrenalina também promove a liberação e quebra de nutrientes de gordura que irão viajar para o fígado para ser convertida em cetonas e açúcar
Cortisol
O cortisol é um tipo de hormona esteróide que é secretada a partir da glândula adrenal do corpo. Ele funciona para tornar as células musculares e gordas resistentes à ação da insulina, e que também aumenta a produção de glicose no fígado. Cortisol irá contrabalançar a ação da insulina, mas sob stress, os níveis de cortisol podem aumentar até o ponto onde você se torna resistente à insulina. Para as pessoas com diabetes tipo 1, o que exigirá insulina extra, a fim de controlar os seus níveis de açúcar no sangue.
Hormônio do Crescimento
A hormona de crescimento é libertada a partir da glândula pituitária do cérebro, e funciona de forma semelhante ao cortisol. O hormônio do crescimento vai trabalhar para contrabalançar o efeito da insulina nas células musculares e adiposas. No entanto, quando os níveis de hormona de crescimento são demasiado elevados, resistência à insulina pode resultar.
(1)
O peptídeo-C faz parte da molécula de pró-insulina, que, ao ser clivada, dá origem a esse peptídeo e à insulina, ambos armazenados e secretados juntos nas células-beta pancreáticas, entendeu-se que seria apenas um resíduo resultante da formação da insulina. Com isso, a dosagem de peptídeo-C consagrou-se como forma indireta para detectar e quantificar a produção de insulina.
Os caminhos pelos quais esse peptídeo elícita suas ações biológicas, resultando em consequências positivas, como coadjuvante no controle do diabetes para a prevenção de complicações. Destacam-se seus papeis como anti-inflamatório, mediando a inibição da NF-κβ, além da expressão e concentração de uma série de citocinas, quimiocinas e peptídeos pró-inflamatórios; antioxidante, reduzindo a formação de espécies reativas de oxigênio; anti-apoptótico, inibindo a caspase 3 e estimulando a proteína Bcl-2; na melhoria do fluxo sanguíneo em diferentes tecidos, como resultado da ativação e indução da expressão do eNOS; e na correção da bomba Na+/K+também em diferentes tecidos. Algumas das consequências disso são: efeitos citoprotetores sobre células vasculares e neuroprotetores, interferência na formação de lesão aterosclerótica, prevenção e reversão parcial do déficit na condução nervosa, melhora da sensibilidade periférica e da disfunção erétil, redução da hiperfiltração glomerular e da excreção de albumina, prevenção de vazamento na retina de animais com diabetes induzido, entre outras.
Ao mesmo tempo, é bastante evidente que se um dia o peptídeo-C passar a fazer parte do tratamento do diabetes, especialmente do tipo 1, será de forma complementar ao tratamento com insulina, alimentação saudável e exercício físico. Portanto, as consequências reconhecidamente positivas do tratamento atual não poderão ser substituídas, mas talvez ampliadas. Além disso, os autores deixam claro que os efeitos do peptídeo-C não se intensificam com aumento progressivo da dose, sendo máximos ao se atingir a concentração fisiológica, quando acontece a saturação dos receptores. Sugerem, ainda, mais estudos para se entender os mecanismos e efeitos sobre diferentes tecidos e, especialmente, os resultados nos casos de complicações em estágios mais avançados.
(2)
Estudos de menor porte utilizando ensaios ultrassensíveis de peptídeo-C sugere que a secreção endógena de insulina pode frequentemente ser detectável em pacientes com diabetes tipo 1 (DM1) de longa duração, mas esses estudos não usam amostras representativas. Para este estudo, os pesquisadores utilizaram o parâmetro estimulado do coeficiente peptídeo-C/creatinina para avaliar os níveis reais de peptídeo-C numa ampla população de pacientes com DM1.
Secretado em concentrações equimolares às da PL insulina, o peptídeo C consiste no fragmento liberado quando a proinsulina sofre clivagem, dando origem ao hormônio propriamente dito. Dessa forma, serve como marcador de reserva funcional de células beta em diabéticos do tipo 1, nos quais a dosagem de insulina pode ficar prejudicada devido ao consequente aparecimento de anticorpos endógenos, assim como no diagnóstico da hipoglicemia factícia, em que caracteristicamente não se detecta o peptídeo C. Ademais, níveis elevados desse fragmento, concomitantes com quadros de hipoglicemia, podem ser encontrados em casos de insulinoma.
Como a dosagem dessa substância não sofre influência da presença de anticorpos anti-insulina nem da própria insulina, seu nível em jejum, ou depois de qualquer teste de estímulo, tem sido utilizado para o seguimento da história natural da função das células beta no diabetes mellitus tipo 1 (DM1), após o início do uso do hormônio exógeno.
NOVA SÍNTESE:
Peptídeo C --> metabólito resultante da transformação da pró-insulina em insulina
Em DM2 - auto peptídeo C
Insulina- superprodução inicial com queda subsequente devido à degradação de células beta
Glucagon- elevada taxas devido o metabolismo defasado da glicose
REFERÊNCIAS:
(1)Diabetes Health, Stephanie Clarke, 2018 - https://www.diabeteshealth.com/the-link-between-diabetes-and-hormones/
(2) Dr. Mark Barone [Doutor em Fisiologia Humana (ICB/USP), Especialista em Educação em Diabetes (ADJ-IDF-SBD, UNIP e IDC)] - Peptídeo-C, de inútil a Pop Star, 2015 - https://www.diabetes.org.br/publico/colunistas/89-dr-mark-barone/1098-peptideo-c-de-inutil-a-pop-star
6. Quais são os fatores de risco para DM tipo II?
Ter tido diabetes gestacional, ou ter dado a luz a pelo menos um filho pesando mais que 4 kg.
Ter pressão 140/90 ou maior.
Ter níveis de colesterol anormais: HDL menor que 35 e triglicérides maior que 250.
Praticar exercícios físicos menos que três vezes por semana.
Ter síndrome dos ovários policísticos.
Histórico de doença cardiovascular.
Pressão alta
Colesterol alto
Idade acima de 45 anos
Genética ( histórico familiar)
Sedentarismo
Obesidade
Distribuição da gordura ( Se o organismo armazena a gordura primariamente no abdômen, o risco de diabetes tipo 2 é maior do que se o organismo a armazenasse em outros locais)
Pré-diabetes ( O pré-diabetes é uma condição em que sua glicemia é maior que o normal, porém, não alta o suficiente para ser classificada como diabetes. Se não tratado, o pré-diabetes pode levar ao risco de desenvolver diabetes tipo 2)
Diabetes gestacional ( Caso uma mulher tenha desenvolvido diabetes gestacional durante a gestação, seu risco de desenvolver diabetes tipo 2 posteriormente aumenta. Caso você tenha dúvidas com relação ao seu risco de diabetes tipo 2 ou aos sintomas apresentados, fale com um profissional de saúde )
NOVA SÍNTESE:
Histórico familiar
Síndrome metabólica
Pré-diabetes
Obesidade
Tabagismo
Transtornos psiquiátricos
Mãe cujo filho nasce com peso maior que 4kg
Apneia do sono
Colesterol alto
Diabetes gestacional
Uso de corticoides
Doença renal crônica
Síndrome dos ovários policístico
Etilismo
REFERÊNCIAS:
Abbot - 2017 - https://www.lifetothefullest.abbott/pt/articles/entenda-os-fatores-de-risco-do-diabetes-tipo-2-e-previna-se.html
MALFACINI, Luciana - 2016 - CENTRO UNIVERSITÁRIO IBMR – LAUREATE INTERNATIONAL UNIVERSITIES CURSO DE NUTRIÇÃO - https://www.ibmr.br/files/tcc/diabetes-mellitus-fatores-de-risco-prevencao-e-tratamento-luciana-de-o-malfacini.pdf


Comentários